


Mujer de 50 años con antecedentes personales de histerectomía por miomatosis uterina (1997), carcinoma renal de conductos colectores que fue tratado mediante nefrectomía radical derecha (2005) y cistoadenoma mucinoso ovárico izquierdo para el que se realizó una anexectomía (2010). Con respecto a sus antecedentes familiares llamó la atención que un hermano y una hermana habían fallecido por carcinoma renal.
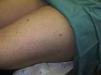
Acudió a la consulta de dermatología por la presencia de múltiples lesiones cutáneas en el tronco y la raíz de los miembros, dolorosas al tacto y con las bajas temperaturas, que habían ido apareciendo progresivamente tras el diagnóstico del cáncer renal (fig. 1).
Exploración física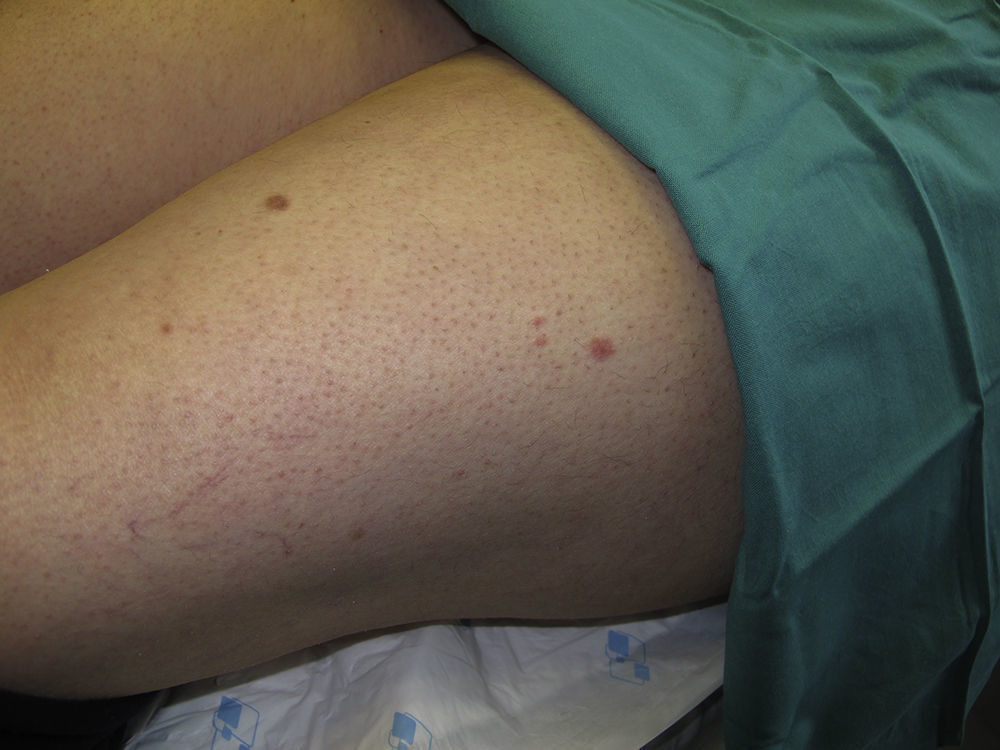
Se observaban lesiones nodulares eritematosas de predominio en el tronco y la raíz de los miembros, en número de 15 a 20 en total.
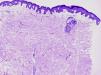
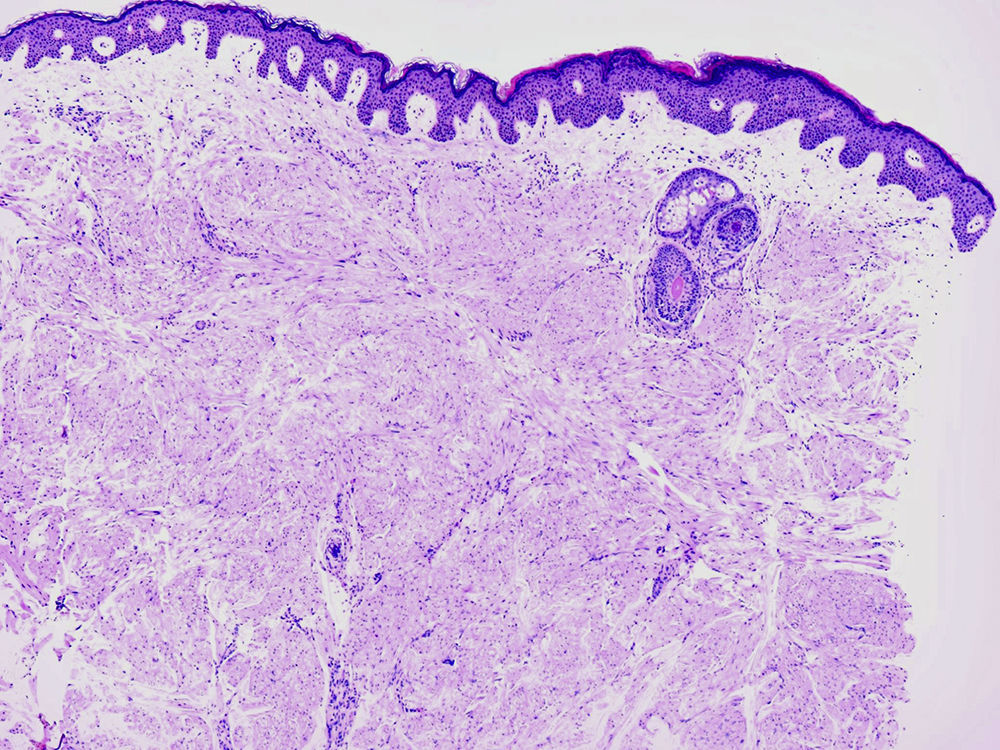
HistopatologíaEl estudio histológico reveló una tumoración no encapsulada localizada en la dermis, compuesta por haces de músculo liso organizados en un patrón entrelazado. Las células presentaban un abundante citoplasma eosinófilo con núcleos alargados de terminaciones romas (fig. 2).
¿Cuál es su diagnóstico?
DiagnósticoLeiomiomatosis hereditaria y carcinoma de células renales (LHCCR).
EvoluciónTras años de seguimiento el proceso renal se ha mantenido sin signos de recidiva ni de enfermedad a distancia. Las lesiones cutáneas han continuado aumentando progresivamente en número y tamaño durante años. Se remitió para estudio genético propio y de familiares.
TratamientoSe realizó exéresis quirúrgica de las lesiones cutáneas más molestas.
ComentarioLa LHCCR es una entidad poco frecuente que se caracteriza por la existencia de leiomiomas, cutáneos y uterinos, asociados a carcinoma de células renales1. Este último se suele presentar de forma unilateral (lo que contrasta con otros síndromes de cáncer renal hereditario como el de Von Hippel Lindau o el de Birt-Hogg-Dubé), tiende a ser más agresivo y su forma histológica predominante es el carcinoma papilar tipo 2, aunque hay casos descritos de carcinomas de conductos colectores, de células claras y mixtos papilares y túbulo-papilares. Es raro que los piloleiomiomas cutáneos y los leiomiomas uterinos degeneren a variantes malignas, aunque hay algunos casos descritos y se ha observado que tienden a aparecer a edades más tempranas1. Además, se ha descrito la asociación entre la mutación del gen responsable con otras neoplasias como las de mama, próstata y hematológicas.
No debe confundirse la LHCCR con el síndrome de Reed, en el que se asocian leiomiomas cutáneos y uterinos, pero sin relación con el cáncer renal2.
En su etiología se ha implicado la afectación del gen 1q42.3-q43, que se hereda de forma autosómica dominante y que codifica para la enzima fumarato hidratasa, implicado en el sistema de supresores de tumores integrado en el ciclo de Krebs. Su alteración conduce a un cúmulo de fumarato, que va a inhibir la enzima que hidroliza el factor inducible por hipoxia (HIF), elevando sus niveles y, así, la transcripción de genes implicados en la carcinogénesis3,4.
Con respecto a la indicación y elección del tratamiento más adecuado de los piloleiomiomas cutáneos siempre dependerá del número, tamaño, localización y sintomatología5. De este modo, en pacientes con un gran número de ellos deberán tratarse aquellos que produzcan más molestias, en pacientes con piloleiomiomas de gran tamaño evitaremos exéresis quirúrgicas deformantes, mientras que en piloleomiomas únicos o escasos optaremos por la cirugía.
En nuestro caso, dado el número, tamaño y localización de los piloleiomiomas, optamos por la exéresis quirúrgica como medida más adecuada.
El conocimiento de esta enfermedad por el dermatólogo va a permitir un diagnóstico más precoz, que deberá ser confirmado con un estudio genético, tanto del paciente como de los familiares disminuyendo, de este modo, la morbimortalidad de los afectados6.
Conflicto de interesesLos autores declaran que no tienen ningún conflicto de intereses.


