









Las herramientas para el diagnóstico en las epidermólisis ampollosas (EA) han tenido un gran avance desde que Hintner et al, introdujeron el mapeo antigénico como prueba diagnóstica en este grupo de genodermatosis. La utilización de anticuerpos monoclonales/policlonales dirigidos contra algunas de las proteínas específicas que conforman la epidermis y la membrana basal epidérmica, han servido para clasificar los 4 tipos de epidermólisis ampollosa y subclasificar todas sus variantes. Ante la presencia de un recién nacido con ampollas surgen diagnósticos diferenciales múltiples, en donde la microscopia de luz orienta el diagnostico de epidermólisis ampollosa. Sin embargo, el mapeo por inmunofluorescencia y la microscopia electrónica permiten confirmar y clasificar a las epidermólisis ampollosas congénitas.
En este artículo, se explica la importancia y metodología para desarrollar la técnica de mapeo antigénico por inmunofluorescencia, con el propósito de clasificar y subclasificar las epidermólisis ampollosas.
The tools for diagnosis of epidermolysis bullosa have advanced greatly since Hintner's group introduced antigen mapping as a diagnostic test for this family of genodermatoses. Monoclonal or polyclonal antibodies raised against some of the specific proteins found in the epidermis and basement membrane of the epidermis have allowed 4 types of epidermolysis bullosa de be identified and all variants to be classified. When a newborn baby presents with blisters, many conditions are implicated in the differential diagnosis. Examination under an optical microscope can suggest epidermolysis bullosa, but immunofluorescence mapping and electron microscopy are required for confirmation of the diagnosis and further classification of congenital epidermolysis bullosa. This article explains the importance of immunofluorescence antigen mapping and describes the methods employed for classification and subclassification of epidermolysis bullosa.
El termino epidermólisis ampollosa (EA) (EB, por sus siglas en ingles Epidermolysis Bullosa) empleado por primera vez por Koebner en 1886 describe un grupo de enfermedades genéticas, clínicamente heterogéneas, caracterizadas por la aparición de ampollas, erosiones y úlceras en piel y/o mucosas, secundarias al mínimo traumatismo y/o de aparición espontanea, razón por la que también se les denomina enfermedades mecano-ampollosas1.
De acuerdo al fenotipo, modo de herencia y genotipo, actualmente han sido descritos más de 30 subtipos de EA. No obstante, la enfermedad se divide en tres tipos principales de acuerdo al nivel en que se encuentran las ampollas2,3, determinados por inmunofluorescencia y/o microscopia electrónica (ME): epidermólisis ampollosa simple (EAS), de unión (EAU) y distrófica (EAD). En las formas simples (EBS, epidermolysis bullosa simplex) las ampollas se encuentran en la epidermis (epidermolíticas), en la EA de unión (JEB, juntional epidermolysis bullosa) es posible observar la separación de la lamina lucida de la unión dermo-epidérmica y en las formas distróficas de EA (DEB, dystrophic epidermolysis bullosa) las ampollas afectan la dermis papilar (dermolíticas)4,5. De acuerdo a la nueva clasificación publicada en 2008, por el consenso internacional para el diagnóstico de EA, ha sido añadido un cuarto tipo mixto (síndrome de Kindler), basado en los diferentes niveles en los que se presentan las ampollas en la piel afectada6 (fig. 1).
.")
El desafío en el diagnóstico de los tipos de EA es particularmente difícil, si solo se cuenta con la presentación clínica del paciente y aun más laborioso en ausencia de historia familiar del paciente7. El recién nacido es propenso a desarrollar ampollas y erosiones en respuesta a agentes externos como calor, irritantes químicos y traumatismos8,9 (fig. 2). A este hecho se añade que durante el periodo neonatal se presentan la mayoría de las enfermedades caracterizadas por fragilidad en la piel, circunstancia que nos plantea un amplio espectro de diagnósticos diferenciales de EA congénita, desde enfermedades transitorias benignas, hasta procesos mutilantes que comprometen la vida del paciente. El diagnóstico diferencial debe comprender las siguientes enfermedades: ampollas por succión, eritrodermia ictiosiforme congénita ampollosa, impétigo ampolloso, síndrome de piel escaldada por estafilococo, herpes simple neonatal, varicela zoster intrauterina, incontinencia pigmentaria (síndrome de Bloch-Sulzberger), aplasia cutis, hipoplasia focal dérmica (síndrome de Goltz), porfiria eritropoyética congénita (enfermedad de Gunther), lupus eritematoso neonatal, ectima gangrenoso, infección por Aspergillus y enfermedades ampollosas autoinmunes como, pénfigo o penfigoide gestationis8–11.

Por otro lado, en infantes y adultos, la epidermólisis ampollosa congénita presenta características clínicas que recuerdan a la mastocitosis ampollosa, nécrolisis epidérmica tóxica o algunas enfermedades autoinmunes como penfigoide ampolloso, penfigoide cicatrizal, enfermedad IgA lineal, o EA adquirida9–11.
Una vez excluidas estas patologías mediante una anamnesis detallada, junto a la exploración física completa, y la realización de cultivos de las lesiones con tinciones de Gram y Giemsa para infecciones bacterianas y frotis de Tzanck en los casos de sospecha de infección viral o KOH en micosis, el diagnóstico de EA deberá sospecharse. En estos casos, realizaremos una biopsia de piel, teñida con hematoxilina y eosina así como mapeo antigénico por inmunofluorescencia y/o para microscopía electrónica. Mediante estas dos últimas técnicas se pretende determinar el plano de clivaje o sitio de formación de las ampollas en la epidermis o membrana basal epidérmica9–13.
La microscopia de luz tiene poco valor diagnóstico para la enfermedad, debido a que en la mayoría de los casos únicamente se observa una ampolla subepidérmica pobre en células inflamatorias, indistintamente del tipo de EA. Sin embargo en algunos casos de EAS es posible visualizar ampollas intraepidérmicas en el estrato basal, secundarias a la citólisis de los queratinocitos basales14 (fig. 3).
 Ampolla sub epidérmica carente de células inflamatorias característica de cualquier forma de epidermólisis ampollosa H y E 20×. b) Ampolla basal característica de la EA simple H y E 20×. c) Dos queratinocitos remanentes adheridos a la membrana basal en EA simple (*) H y E 60×.")
Para llevar a cabo el diagnóstico y la clasificación precisa de la enfermedad, la primera herramienta de laboratorio utilizada con éxito fue la ME y durante muchos años ha sido la técnica diagnóstica más empleada, ya que permite visualizar detalladamente la estructura de las células, organelas, citoplasma y especialmente los tonofilamentos de citoqueratinas, hemidesmosomas y fibrillas de anclaje, involucrados en esta patología15. Además, permite observar el sitio de separación o formación de la ampolla en los queratinocitos basales y/o membrana basal. Sin embargo, la ME es una herramienta costosa, consume tiempo y principalmente es necesario contar con un especialista en la técnica con experiencia en la enfermedad.
Por esta razón la interpretación de los resultados de la ME, pueden ser en ocasiones imprecisos y solo existen pocos laboratorios con apropiada experiencia y habilidades para el análisis e interpretación de las muestras de los pacientes con EA congénita5,7. En 1981 Hintner et al, describieron por primera vez el método de mapeo antigénico por inmunofluorescencia para la enfermedad, el cual se basa en la detección de proteínas estructurales de la epidermis y/o la unión dermo-epidérmica usando anticuerpos policlonales y/o monoclonales5. Con este método se busca determinar el sitio de clivaje y la localización de la ampolla, al exhibir un antígeno determinado (como por ejemplo, colágeno tipo iv) en una ampolla «natural» o inducida. Esta técnica, dependiendo de los anticuerpos utilizados, permite comprobar la expresión normal, reducción o ausencia de las proteínas estructurales. Actualmente existen varios anticuerpos que reconocen las proteínas estructurales de los queratinocitos y/o de la unión dermo-epidérmica en la membrana basal, los cuales se conoce que están involucrados en la patogénesis de la enfermedad.
Afortunadamente algunos de estos anticuerpos están disponibles comercialmente en todo el mundo para el desarrollo del método de inmunofluorescencia cutánea6,16,17. El mapeo por inmunofluorescencia ha desplazado a la ME y actualmente es el primer examen de laboratorio empleado para hacer el diagnóstico de EA y distinguir los diferentes tipos de la enfermedad. Además es la base para distinguir las proteínas blanco del análisis de mutaciones6,15.
BiopsiaEl sitio adecuado para tomar la biopsia de piel en un paciente con sospecha de EA congénita es preferentemente la zona perilesional sana de una ampolla reciente (fig. 4). En los casos en que no se encuentre una ampolla reciente debe friccionarse la piel sana con un borrador durante 2min con el fin de producir una ampolla nueva. El propósito de tomar una biopsia reciente o inducida es evitar los cambios en las proteínas estructurales secundarios a la propia herida y al proceso inherente de cicatrización5–7,18.
Fijación y almacenamiento de la muestra
La muestra de piel debe ser inmediatamente colocada en solución de Michel descrita originalmente por Michel et al en 1973 y modificada por Vaughan et al en 1995 (2,5ml de amortiguador de citratos a un pH de 7,4, con una concentración de 1M, 5ml de sulfato de magnesio a una concentración de 0,1M, 5ml de N-etilmaleimida a una concentración de 0,1M, 55g de sulfato de amonio, diluido en 87,5ml de agua, para un volumen total de 100ml ajustado a un ph de 7,4 con un 1M de hidróxido de sodio)18–20. En este medio las muestras pueden ser almacenadas durante 28 días a temperatura ambiente para ser enviadas a un laboratorio especializado en cualquier parte del mundo para realizar el mapeo antigénico. De forma previa a cortar la muestra, esta debe ser lavada en una solución salina amortiguada por fosfatos por varias horas para mejorar la sensibilidad diagnóstica. Después, las muestras pueden ser cortadas en criostato para posteriormente teñirlas21–23.
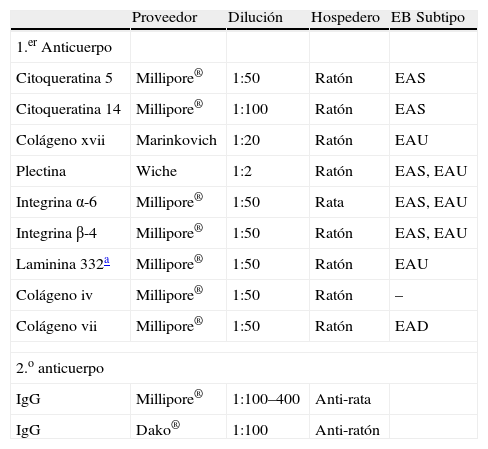
AnticuerposLos anticuerpos primarios para realizar la técnica de mapeo por inmunofluorescencia cutánea tienen diferentes orígenes animales (rata, ratón, conejo), los cuales se fijan a las proteínas estructurales específicas en la piel que deseamos estudiar. Los anticuerpos que podemos utilizar para el mapeo antigénico están dirigidos contra citoqueratina 5, citoqueratina 14, plectina, integrina α6 y β4, colágeno tipo xvii (180kD PBAG2), laminina 332 (anteriormente conocida como laminina 5) con sus tres cadenas α3, β3, γ2 y finalmente colágeno tipo vii24,25. Para una mejor visualización del nivel de la ampolla, especialmente en pacientes con EA distrófica, se utilizan adicionalmente anticuerpos contra colágeno tipo iv (presente en la lamina densa de la unión dermo-epidérmica) (tabla 1).
Lista de anticuerpos y diluciones utilizadas actualmente en inmunofluorescencia para el diagnóstico de epidermólisis bullosa
| Proveedor | Dilución | Hospedero | EB Subtipo | |
| 1.er Anticuerpo | ||||
| Citoqueratina 5 | Millipore® | 1:50 | Ratón | EAS |
| Citoqueratina 14 | Millipore® | 1:100 | Ratón | EAS |
| Colágeno xvii | Marinkovich | 1:20 | Ratón | EAU |
| Plectina | Wiche | 1:2 | Ratón | EAS, EAU |
| Integrina α-6 | Millipore® | 1:50 | Rata | EAS, EAU |
| Integrina β-4 | Millipore® | 1:50 | Ratón | EAS, EAU |
| Laminina 332a | Millipore® | 1:50 | Ratón | EAU |
| Colágeno iv | Millipore® | 1:50 | Ratón | – |
| Colágeno vii | Millipore® | 1:50 | Ratón | EAD |
| 2.o anticuerpo | ||||
| IgG | Millipore® | 1:100–400 | Anti-rata | |
| IgG | Dako® | 1:100 | Anti-ratón | |
Todos los primeros anticuerpos son de clase IgG y la mayoría de ellos son monoclonales y están desarrollados en ratón. Por lo tanto, el segundo anticuerpo debe estar dirigido hacia el anticuerpo IgG, normalmente conjugado con una partícula de fluorescencia isotiocianato (FITC). Una excepción es el anticuerpo contra la integrina α6, el cual se desarrolla en rata, por lo que el segundo anticuerpo es de tipo IgG anti-rata. La partícula FITC se conjuga con el segundo anticuerpo y produce una señal específica de fluorescencia en un rango de 450–490nm, lo que permite la visualización del anticuerpo específico unido a la proteína en estudio que se desea visualizar en el microscopio de fluorescencia.
Dependiendo de la frecuencia de utilización de la inmunofluorescencia y la dilución final necesaria para la técnica, los anticuerpos deben ser almacenados en cantidades que permitan la preparación en fresco del total de las muestras en cada sesión, con la finalidad de mantener los anticuerpos en congelación hasta la fecha de caducidad y evitar contaminación22.
Técnica de tinciónDurante la técnica de tinción para el mapeo por inmunofluorescencia se recomienda utilizar de 3 a 4 cortes de cada muestra, cada uno de 4–6μ de grosor, así como 1–2 cortes de piel, provenientes de pacientes sanos que se utilizaran como controles positivos (fig. 5). Esto permite obtener un punto de referencia de la inmunofluorescencia de las proteínas estructurales en estudio, para posteriormente comparar la inmunoreactividad de las proteínas del control sano con la inmunoreactividad de las proteínas de los pacientes con EA.

De acuerdo al número de anticuerpos a determinar se preparan igual número de laminillas para la tinción de las posibles proteínas involucradas. Dos de estas laminillas sirven como controles negativos, con la finalidad de evaluar y excluir la inmunoreactividad no especifica causada por el segundo anticuerpo. Las laminillas se incuban en solución salina amortiguada por fosfatos (SSAF) con el primer anticuerpo correspondiente, para ser incubadas con el segundo anticuerpo con las partículas de FITC contra ratón conjugado o contra rata. Cada laminilla es incubada con la dilución apropiada para el primer anticuerpo (SSAF en el caso del control negativo) durante 30min en una cámara húmeda a temperatura ambiente. Después, las muestras se lavan en SSAF en dos ocasiones por 15min, y con la dilución apropiada del segundo anticuerpo se incuban por 30min en las mismas condiciones.
Posteriormente, las muestras deben lavarse nuevamente dos veces en SSAF por 15min, para inmediatamente cubrir con glicerol y cubreobjetos. Finalmente en el microscopio de fluorescencia, se observa y se toma fotografías del patrón y reactividad de la fluorescencia, para ser archivadas en el expediente de cada paciente. Esta técnica logra ser estable por varias semanas cuando se mantienen en refrigeración a 4°C.
Patrones de tinciónEn el mapeo antigénico por inmunofluorescencia es posible visualizar la localización y expresión de las proteínas estructurales involucradas en la patogénesis de las diferentes formas de la enfermedad, así como el posible sitio de separación y/o formación de la ampolla. Sin embargo, la intensidad de la tinción de las proteínas es influenciada por varios factores como son la parte del cuerpo de la que se obtuvo la piel (normal o piel afectada), la exposición solar previa, así como y la edad de los pacientes. Para realizar una adecuada correlación entre piel afectada y el control sano, es indispensable tomar cortes de piel de pacientes de la misma edad y partes del cuerpo que los controles. Además el tiempo de almacenaje en la solución de Michel (preferentemente no más de 28 días), las condiciones de transporte (altas temperaturas, congelación-descongelación) o las condiciones de almacenaje de los anticuerpos pueden influenciar los resultados de la técnica de inmunofluorescencia. Para evaluar los resultados de la tinción es útil hacer una escala del 1–4 de acuerdo a la intensidad de la reacción antígeno-anticuerpo, donde (+) es intensidad dudosa, (++) intensidad débil, (+++) moderada intensidad, (++++) muy brillante. Como se mencionó con anterioridad, es de gran importancia tener una ampolla en la biopsia, ya que esto podrá permitir visualizar el nivel en la piel en la que aparece la separación.
En el caso de EAS, al utilizar por ejemplo el anticuerpo para el colágeno iv (presente en la lamina densa de la unión demo-epidérmica) se encuentran ampollas intraepidérmicas, localizadas en el nivel de los queratinocitos basales: en estos casos observaremos que la fluorescencia, situada en la lámina densa, permanece en el suelo de la ampolla junto a restos de queratinocitos basales unidos a la membrana basal (fig. 6a).
 Ampolla epidérmica en zona de citoqueratina 5, 14, plectina y integrinas α6β4 (20×), b y c) ampolla en la lamina lúcida (20×) (citoqueratina 5 y laminina 332), d) ampolla en dermis papilar debajo de lamina lúcida (20×) (colágeno vii). *Ampollas")
Cuando estudiamos lesiones de EA de unión suelen utilizarse en el mapeo antigénico anticuerpos frente a colágeno iv y lamininas en las técnicas de inmunofluorescencia. En estos casos observaremos que la fluorescencia se encuentra en el piso de la ampolla (fig. 6b y c), aunque en el caso de las citoqueratinas el marcaje únicamente se encuentra en el techo de la ampolla (fig. 6b).
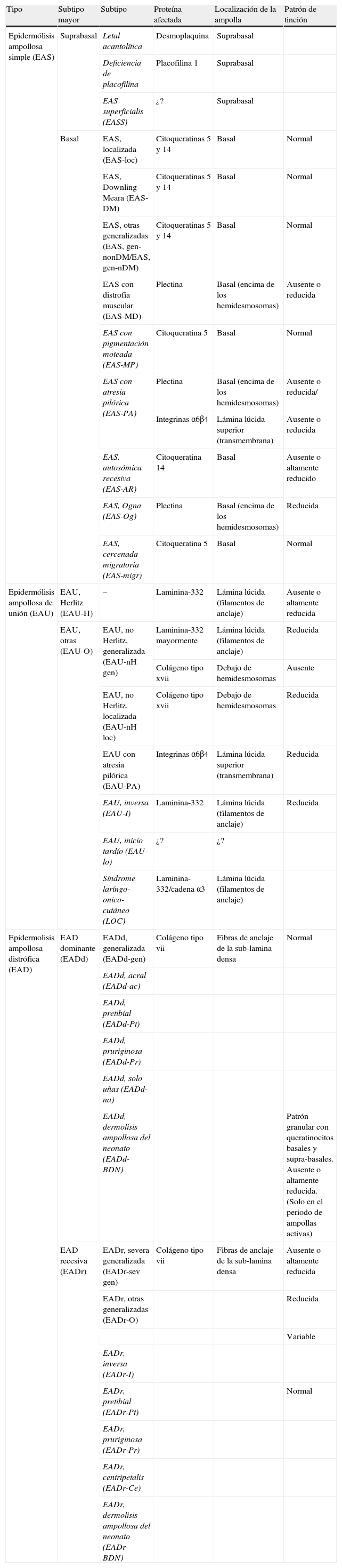
Finalmente, en los casos de EA distróficas los estudios de mapeo suelen realizarse con anticuerpos dirigidos frente a citoqueratinas, colágeno iv y lamininas. De esta manera, observaremos que la fluorescencia se localiza solo en el techo de la ampolla26 (fig. 6d). En cada tipo y subtipo de la enfermedad se pueden distinguir diferentes proteínas alteradas de la piel. Por otro lado la expresión de estas proteínas, puede llegar a ser igual a los controles, reducida en intensidad o completamente ausente (tabla 2).
Alteraciones antigénicas en la epidermólisis ampollosa
| Tipo | Subtipo mayor | Subtipo | Proteína afectada | Localización de la ampolla | Patrón de tinción |
| Epidermólisis ampollosa simple (EAS) | Suprabasal | Letal acantolítica | Desmoplaquina | Suprabasal | |
| Deficiencia de placofilina | Placofilina 1 | Suprabasal | |||
| EAS superficialis (EASS) | ¿? | Suprabasal | |||
| Basal | EAS, localizada (EAS-loc) | Citoqueratinas 5 y 14 | Basal | Normal | |
| EAS, Downling-Meara (EAS-DM) | Citoqueratinas 5 y 14 | Basal | Normal | ||
| EAS, otras generalizadas (EAS, gen-nonDM/EAS, gen-nDM) | Citoqueratinas 5 y 14 | Basal | Normal | ||
| EAS con distrofia muscular (EAS-MD) | Plectina | Basal (encima de los hemidesmosomas) | Ausente o reducida | ||
| EAS con pigmentación moteada (EAS-MP) | Citoqueratina 5 | Basal | Normal | ||
| EAS con atresia pilórica (EAS-PA) | Plectina | Basal (encima de los hemidesmosomas) | Ausente o reducida/ | ||
| Integrinas α6β4 | Lámina lúcida superior (transmembrana) | Ausente o reducida | |||
| EAS, autosómica recesiva (EAS-AR) | Citoqueratina 14 | Basal | Ausente o altamente reducido | ||
| EAS, Ogna (EAS-Og) | Plectina | Basal (encima de los hemidesmosomas) | Reducida | ||
| EAS, cercenada migratoria (EAS-migr) | Citoqueratina 5 | Basal | Normal | ||
| Epidermólisis ampollosa de unión (EAU) | EAU, Herlitz (EAU-H) | – | Laminina-332 | Lámina lúcida (filamentos de anclaje) | Ausente o altamente reducida |
| EAU, otras (EAU-O) | EAU, no Herlitz, generalizada (EAU-nH gen) | Laminina-332 mayormente | Lámina lúcida (filamentos de anclaje) | Reducida | |
| Colágeno tipo xvii | Debajo de hemidesmosomas | Ausente | |||
| EAU, no Herlitz, localizada (EAU-nH loc) | Colágeno tipo xvii | Debajo de hemidesmosomas | Reducida | ||
| EAU con atresia pilórica (EAU-PA) | Integrinas α6β4 | Lámina lúcida superior (transmembrana) | Reducida | ||
| EAU, inversa (EAU-I) | Laminina-332 | Lámina lúcida (filamentos de anclaje) | Reducida | ||
| EAU, inicio tardío (EAU-lo) | ¿? | ¿? | |||
| Síndrome laríngo-onico-cutáneo (LOC) | Laminina-332/cadena α3 | Lámina lúcida (filamentos de anclaje) | |||
| Epidermolisis ampollosa distrófica (EAD) | EAD dominante (EADd) | EADd, generalizada (EADd-gen) | Colágeno tipo vii | Fibras de anclaje de la sub-lamina densa | Normal |
| EADd, acral (EADd-ac) | |||||
| EADd, pretibial (EADd-Pt) | |||||
| EADd, pruriginosa (EADd-Pr) | |||||
| EADd, solo uñas (EADd-na) | |||||
| EADd, dermolisis ampollosa del neonato (EADd-BDN) | Patrón granular con queratinocitos basales y supra-basales. Ausente o altamente reducida. (Solo en el periodo de ampollas activas) | ||||
| EAD recesiva (EADr) | EADr, severa generalizada (EADr-sev gen) | Colágeno tipo vii | Fibras de anclaje de la sub-lamina densa | Ausente o altamente reducida | |
| EADr, otras generalizadas (EADr-O) | Reducida | ||||
| Variable | |||||
| EADr, inversa (EADr-I) | |||||
| EADr, pretibial (EADr-Pt) | Normal | ||||
| EADr, pruriginosa (EADr-Pr) | |||||
| EADr, centripetalis (EADr-Ce) | |||||
| EADr, dermolisis ampollosa del neonato (EADr-BDN) |
Fuentes: Fine JD et al, Yiasemides et al6,7.
En pacientes con EAS (fig. 7a) las principales proteínas involucradas en la patología de la enfermedad son las citoqueratinas 5 y 14, plectina, integrina α6 e integrina β4. La alteración en alguna de estas proteínas promueve la formación de ampollas intraepidérmicas, por citólisis de los queratinocitos basales (fig. 7b–d). En general las proteínas son expresadas con una intensidad similar a los controles, excepto en la EAS autosómica recesiva26, donde los pacientes tienen ausencia de citoqueratina 14.
 Manifestaciones clínicas de un pacientes con EAS. b) Control positivo de piel sana teñido para citoqueratina 5 (20×) c y d) citoqueratina 5 y 14 las cuales muestran ampollas intraepidérmicas basales (20×).")
En pacientes con EAS con ditrofia muscular (EAS-MD) la plectina esta generalmente ausente y en raros casos como en EAS Ogna la plectina se encuentra marcadamente reducida. En los pacientes con atresia pilórica (EAS o EAU) la plectina, así como las integrinas α6β4 se encuentran reducidas o ausentes27,28.
Epidermólisis ampollosa de unión (EAU)Las proteínas blanco principales en EAU son el colágeno tipo xvii (PBG2) y la laminina 332. Estas proteínas pueden ser expresadas normalmente, reducidas o ausentes, al compararlas con el control sano. En los casos con ampollas en la unión dermoepidérmica, la inmuno-reactividad aparece en el techo o en el piso de las ampollas dependiendo del tipo de EAU29,30 (fig. 8).
 Pacientes con EAU-nH generalizada; ampollas hemorrágicas, onicodistrofia, ampollas tensas y costras hemorrágicas. e y f) laminina 332 control (20×). f) Laminina 332 ausente característico de EAU-Herlitz (20×) l. g) Laminina-5 reducida en intensidad (20×) y h) colágeno tipo vii (20×) de intensidad normal en el piso de la ampolla (flechas rojas) característico de EAU-nH gen.")
a–d) Pacientes con EAU-nH generalizada; ampollas hemorrágicas, onicodistrofia, ampollas tensas y costras hemorrágicas. e y f) laminina 332 control (20×). f) Laminina 332 ausente característico de EAU-Herlitz (20×) l. g) Laminina-5 reducida en intensidad (20×) y h) colágeno tipo vii (20×) de intensidad normal en el piso de la ampolla (flechas rojas) característico de EAU-nH gen.
En los casos del subtipo de EAU-Herlitz la laminina 332 se encuentra ausente en la mayoría de los casos o marcadamente reducida (fig. 8f). En los casos de EAU generalizado no-Herlitz (EAU-nH gen) (fig. 8a–d) las proteínas alteradas son la laminina 332 o el colágeno tipo xvii. En la mayoría de los casos con EAU-nH, la laminina 332 se encuentra reducida (fig. 8g), y en el resto de los pacientes el colágeno tipo xvii está ausente o reducida. Los pacientes que anteriormente se clasificaban como EAU generalizada atrófica benigna (anteriormente GABEB), la reactividad de la laminina 332 permanece similar a los controles.
Epidermólisis ampollosa distrófica (EAD)Los subtipos EA distrófica son causados por mutaciones en el colágeno tipo vii. En los pacientes con el tipo severo generalizado de epidermólisis ampollosa (RDEB, recessive dystrophic epidermolysis bullosa), la inmunofluorescencia al colágeno tipo vii se encuentra ausente, en la mayoría de los casos (fig. 9a–d). En estos casos se utiliza el anticuerpo contra colágeno tipo iv para visualizar el nivel de la ampolla. Una reacción del anticuerpo contra el colágeno tipo iv, en el techo de la separación, indica la presencia de una ampolla en dermis papilar y confirma el subtipo distrófico de EA. Por otro lado, cuando el colágeno iv aparece en el piso de la ampolla deberemos plantear como posibles diagnósticos los subtipos EAU o EAS (figs. 7 y 8).
 EADr-sev gen con pseudosindactilia completa en ambas manos y pies acompañada de contracturas. c y d) Ausencia completa de colágeno tipo vii (20×). e y f) EADr-O con ampollas, erosiones y cicatrices atróficas, en tórax y extremidades superiores. g) Control normal (20×) comparado con, h) una ampolla en dermis papila con colágeno tipo vii marcadamente reducido en intensidad (20×).")
a y b) EADr-sev gen con pseudosindactilia completa en ambas manos y pies acompañada de contracturas. c y d) Ausencia completa de colágeno tipo vii (20×). e y f) EADr-O con ampollas, erosiones y cicatrices atróficas, en tórax y extremidades superiores. g) Control normal (20×) comparado con, h) una ampolla en dermis papila con colágeno tipo vii marcadamente reducido en intensidad (20×).
La expresión del colágeno tipo vii puede estar disminuido o incluso ser normal en el subtipo de EA distrófica recesiva otros generalizados (EADr-O)6 (fig. 9e–h).
ConclusiónEl mapeo antigénico por inmunofluorescencia es el procedimiento de elección para el diagnóstico preliminar y clasificación posterior de los diferentes tipos de EA. Los patrones de inmunofluorescencia, (para determinar el nivel de la formación de ampollas y visualizar la expresión de proteínas específicas) identifican las proteínas involucradas en la fisiopatología de la enfermedad. Aunque la identificación de los genes alterados en cada paciente es el criterio definitivo para el diagnóstico de la enfermedad, el mapeo antigénico permite evaluar en un primer momento la expresión de las proteínas epidérmicas y localizar así la zona de la epidermis alterada. Posteriormente, deberemos realizar estudios moleculares que nos permitan realizar un diagnóstico definitivo al analizar las mutaciones en las proteínas estructuralmente alteradas.
La inmunofluorescencia es la primera herramienta de laboratorio de utilidad para brindar asesoramiento a los padres y pacientes acerca del pronóstico clínico, fenotipo e historia natural de la enfermedad, con la ventaja de obtener el resultado el mismo día que se realiza la biopsia. Además, esta técnica puede realizarse más fácilmente en los hospitales, al no requerir técnicamente tanta preparación como la ME. La disponibilidad de medios de fijación de los tejidos (como la solución de Michel) que permiten además su trasporte a centros especializados en esta patología, puede ayudar a los médicos que atienden a pacientes con EA, para poder realizar un primer diagnóstico de la enfermedad.
FinanciaciónEl presente trabajo fue financiado por la Cátedra de Hematología y Cáncer del Centro de Investigación Clínica. Instituto Tecnológico y de Estudios Superiores de Monterrey, Servicio de Patología, Hospital Universitario «Dr. José E. González» Universidad Autónoma de Nuevo León, DEBRA MEXICO AC, DEBRA AUSTRIA y DEBRA INTERNATIONAL.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


