
























Introducción
Casi todas las anomalías vasculares congénitas afectan a la piel y son evidentes en el momento del nacimiento o en las primeras semanas de vida. Se considera que hasta casi un 12 % de los recién nacidos puede presentar una mancha vascular de nacimiento, aunque la mayor parte de ellas desaparecerá en el primer año1. Durante siglos estas lesiones se han conocido como nevus vasculares cutáneos, atribuyéndose según las diferentes culturas a los «antojos» o excesos en el consumo de frutas rojas por la madre durante el embarazo. En el siglo xix, con los inicios de la histopatología, comenzaron a denominarse «angiomas», aunque a menudo, esta nomenclatura se ha utilizado erróneamente para referirse a diferentes lesiones. Por ejemplo, el término hemangioma, el más significativo, se ha aplicado genéricamente a todo tipo de lesiones vasculares, independientemente de su patogenia, características histológicas o curso clínico.
Aunque inicialmente los dos tipos más frecuentes de manchas vasculares de nacimiento, los hemangiomas y las malformaciones vasculares, pueden ser muy parecidas, son diferentes en su evolución y tratamiento. Los hemangiomas aparecen en las primeras semanas o meses de vida, mientras que las malformaciones vasculares siempre están presentes en el nacimiento, aunque desde el principio no sean aparentes. Los hemangiomas suelen regresar espontáneamente con el paso del tiempo, mientras que las malformaciones vasculares nunca desaparecen y suelen crecer durante toda la vida2. Así, en líneas generales se considera que la mayor parte de los hemangiomas son tumores banales que, salvo ciertas excepciones, no requerirán tratamiento, suponiendo más un problema estético que médico, aunque siempre con una gran repercusión psicológica en el ambiente familiar. Al igual que existen hemangiomas congénitos de desarrollo intrauterino que inician la fase de regresión en el momento del parto y la completarán en pocos meses, también existen algunos hemangiomas con desarrollo normal, que no involucionan y persistirán durante toda la vida. Este grupo, definido inicialmente por Enjolras et al3, y caracterizado clínicamente por una única lesión localizada casi siempre en el polo cefálico e histológicamente por ser GLUT-1 negativo, es el único que podría confundirse con una malformación vascular.
Así, en resumen, salvo la excepción comentada, ninguna lesión vascular de nacimiento presente en un adolescente o adulto debe denominarse hemangioma o angioma, porque realmente se trata de una malformación vascular. La confusión sobre las anomalías vasculares es grande, incluso en la comunidad científica, apreciándose en centros de referencia como más de la mitad de los pacientes remitidos con anomalías vasculares han sido diagnosticados y seguidos de forma incorrecta4. Con razón algunos médicos familiarizados con esta patología definen a los niños con anomalías vasculares como «pacientes nómadas», ya que en función de la localización anatómica y posible repercusión sobre órganos específicos, con el paso del tiempo van visitando diversos especialistas, aportando cada uno un diagnóstico distinto y diferentes criterios terapéuticos. Muchas veces, en el mejor de los casos, el tratamiento y seguimiento consiste en la «foto-terapia», término referido no a las propiedades terapéuticas de la radiación ultravioleta, sino a la realización de fotografías periódicas para valorar la evolución de la lesión, y digo en el mejor de los casos, cuando no se realizan tratamientos desproporcionados o inapropiados por un error diagnóstico.
Por tanto, parece necesario, además de una correcta clasificación de las anomalías vasculares, un enfoque multidisciplinar respecto del seguimiento y las posibilidades terapéuticas de estos pacientes. En el año 1982 Mulliken y Glowacki5 publicaron una clasificación biológica de las lesiones vasculares basada en las características del endotelio predominante; esta clasificación ya es clásica y se revisa cada dos años por la International Society for the Study of Vascular Anomalies (ISSVA). A pesar de ello, actualmente persiste una importante confusión nosológica en este campo; por ejemplo, analizando las últimas ediciones de 5 textos de referencia de medicina genética, se aprecia un empleo inapropiado de los términos hemangioma y malformación vascular6, amén de las confusiones todavía más frecuentes en revistas clínicas7,8.
La ISSVA fue fundada en el año 1992 en Budapest con el objeto de consensuar a profesionales de diversos campos de la medicina que están en contacto con estos pacientes pediatras, dermatólogos, radiólogos intervencionistas, cirujanos plásticos y vasculares, cirujanos pediátricos, otorrinos, oftalmólogos, patólogos y genetistas, entre otros, y tiene el objetivo común de mejorar el conocimiento de la etiopatogenia, diagnóstico y tratamiento de los pacientes con lesiones vasculares. El tratamiento de estas lesiones muchas veces es complejo, y debe ser consensuado por un amplio grupo de especialistas que, trabajando en equipo, consiga soluciones definitivas o paliativas para los pacientes con malformaciones vasculares, limitando la «foto-terapia» sólo a aquellos casos donde realmente, tras un estudio detallado, prime la máxima de «ante todo no hacer daño». Entre todos tenemos la responsabilidad de enseñar e ilusionar a todos los profesionales en contacto con estos pacientes, para que sean capaces de remitirlos a centros de referencia y así favorecer el mejor diagnóstico, tratamiento y seguimiento de las malformaciones vasculares.
Clasificación de las malformaciones vasculares
Hace más de dos décadas Mulliken y Glowacki5 describieron una clasificación biológica de las anomalías vasculares congénitas basada en las características patológicas del endotelio predominante y en la evolución natural (tabla 1). Esta clasificación fue posteriormente redefinida por Mulliken y Young9, en 1996 fue adoptada por la ISSVA y hoy en día es considerada clásica con mínimos cambios (tabla 2). En el año 1988 tiene lugar la denominada clasificación de Hamburgo (posteriormente homologada por la ISSVA), donde la malformación es descrita en relación con el componente predominante de la lesión vascular y entonces clasificada como troncular o extra-troncular, dependiendo del estado embrionario cuando se produjo el defecto en el desarrollo10,11 (tabla 3). Esta clasificación no incluye los hemangiomas ni las malformaciones vasculares linfáticas, pero es útil para realizar un diagnóstico clínico-anatómico, sirve como base para definir el tratamiento de elección y facilita la comunicación entre los diferentes especialistas.



Waner y Suen12 han añadido dos pequeñas modificaciones en la clasificación de Mulliken; por un lado consideran erróneo el término «malformaciones arterio-venosas», y sugieren cambiarlo por el de malformaciones capilares, ya que es en el lecho capilar donde se producen las pequeñas uniones arterio-venosas, siendo todos los demás hallazgos característicos (hipertrofia de la arteria aferente y dilatación del sistema venoso eferente) efecto secundario de lo anterior. Aunque esta primera modificación no altera la clasificación previa, ya que los autores consideran que más que aclarar podría originar confusión, la segunda sí lo hace. Concretamente, introducen el término de malformación vascular venular para denominar a la mancha en vino de Oporto o nevus flammeus, defienden que histológicamente estas lesiones se corresponden con vénulas postcapilares ectásicas del plexo papilar y las subclasifican según el tamaño. De manera que se dividen en:
1. Vasos de entre 50-80 um de diámetro; caracterizadas clínicamente por máculas rosadas.
2. Entre 80-120 um, de coloración más intensa que la anterior.
3. Entre 120-150 um y coloración rojo-violácea.
4. Mayores de 150 um, correspondientes a vasos dilatados que forman nódulos palpables, de aspecto en empedrado y coloración violácea.
A este grupo también pertenecen las denominadas malformaciones vasculares venulares de la línea media, vulgarmente conocidas como mancha salmón, picadura de la cigüeña o beso del ángel.
Por otra parte, otros especialistas13 han realizado una clasificación de las malformaciones vasculares venosas en función de la localización anatómica y características hemodinámicas, que resulta especialmente útil para valorar la eficacia del tratamiento esclerosante. Para definir las características hemodinámicas de la lesión debe realizarse una flebografía, aunque como más adelante veremos quizás en el futuro esta prueba pueda suplirse con una angio-tomografía computarizada (TC). Así, las malformaciones venosas quedan divididas en 4 grupos: a) malformaciones aisladas sin drenaje periférico; b) malformaciones que drenan en venas normales; c) malformaciones que drenan en venas displásicas; y d) ectasias venosas. Las tipo a y b son más fáciles de tratar y tienen una mejor respuesta a la escleroterapia.
Finalmente, la historia natural de una malformación arterio-venosa puede presentar diferentes estadiajes clínicos según el momento evolutivo. Schobinger8 los divide en:
I. Quiescente: se carateriza por una mancha rosada-violácea y la presencia de una derivación arterio-venosa detectable por eco-doppler.
II. Expansivo: igual al estadio I, pero clínicamente pulsátil, siendo evidente la presencia de vasos tensos tortuosos.
III. Destructivo: igual al estadio II, junto a cambios cutáneos distróficos, ulceración, sangrado y dolor continuo.
IV. Descompensado: similar al estadio III, asociado a fallo cardiaco.
Fisiopatogenia
Las malformaciones vasculares son lesiones benignas, no tumorales, presentes siempre desde el nacimiento, pero a veces no visibles hasta semanas o meses después14,15. Su incidencia es de 1,5 %, aproximadamente dos terceras partes son de predominio venoso16, y no distinguen entre sexo y raza. Se trata de errores difusos o localizados del desarrollo embriológico clásicamente atribuidos a mutaciones esporádicas. Sin embargo, algunas evidencias recientes hablan a favor de un posible carácter familiar hereditario. Vikkula et al17,18 han encontrado una mutación caracterizada por un aumento de la actividad del receptor de la tirosín cinasa (tunica intima endothelial kinase-2) Tie-2 en dos familias con malformaciones venosas. El Tie-2 es esencial para el desarrollo temprano de los vasos y un incremento de su actividad puede producir un crecimiento anormal del plexo vascular primario19. El mismo grupo de trabajo también ha definido la implicación de la glomulina en pacientes con malformaciones glomuvenosas20. Por otra parte, se conocen algunas mutaciones genéticas en malformaciones cavernosas cerebrales21 y en malformaciones vasculares combinadas tipo síndrome de Klipel-Trenaunay22 y síndrome de Proteus23. En la misma línea se han descrito alteraciones en la modulación neurológica vascular al menos en dos tipos de malformaciones vasculares24,25; así, las malformaciones venulares son probablemente debidas a una deficiencia relativa o absoluta en la inervación autonómica del plexo venular postcapilar, mientras que las malformaciones arterio-venosas pueden ser debidas a la misma alteración, pero a nivel de los esfínteres precapilares26 (tabla 4).

Tabla 4. Genética de malformaciones vasculares
Para explicar la etiología de una mancha en vino de Oporto se ha acuñado el término de «dermatomo enfermo», por el que una lesión es debida a un defecto total o parcial de la inervación vascular sensorial y autonómica, que origina el crecimiento de los vasos afectos, pudiendo alcanzar el típico aspecto en empedrado. Si el defecto es total, los cambios serán más rápidos en el tiempo. El estudio histológico de estas hipertrofias ha demostrado cambios hamartomatosos difusos afectando, además de la alteración vascular, al tejido conectivo epitelial y a los elementos neurales de la piel, que sugieren la existencia de una mutación somática en el área implicada27. Por otra parte, los esfínteres precapilares son los encargados de regular el flujo sanguíneo a través del nidus. Posiblemente, un defecto de la inervación autonómica o alguna deficiencia de neurorreceptores en esa localización sea el origen de las malformaciones arterio-venosas. En función de si el defecto es total o parcial, variará la edad de presentación y la progresión en la evolución.
Al contrario que los hemangiomas, las malformaciones vasculares no tienen un ciclo de crecimiento y posterior regresión espontánea, sino que persisten para siempre y van creciendo lentamente a lo largo de la vida, a veces en relación con traumatismos, cambios en la presión de la sangre o la linfa, procesos infecciosos, cambios hormonales, etc. En estas lesiones es característico que progresivamente se vaya produciendo una ectasia de las estructuras vasculares, aumentando el diámetro de los vasos pero no el número, expandiéndose por tanto por hipertrofia pero no por hiperplasia, como lo hacen los hemangiomas. En cualquier caso, este concepto clásico es matizable, ya que presenta bastantes excepciones. Así por ejemplo las malformaciones arteriovenosas con frecuencia crecen por hiperplasia y pueden comportarse como auténticos tumores. A pesar de una aparente quiescencia endotelial algunas malformaciones vasculares pueden expandirse rápidamente durante un embarazo, después de un tratamiento quirúrgico o en respuesta a un traumatismo. La patogénesis de las malformaciones vasculares no está bien clarificada, pero su formación y progresión están estrechamente relacionadas con la angiogénesis. La angiogénesis constituye un proceso complejo, regulado por muchos factores angiogénicos, que conlleva la formación de una nueva vasculatura funcional, y que incluye la diferenciación de células endoteliales y murales (pericitos), la proliferación y migración celular y la especificación de vasos arteriales, venosos o linfáticos28. Aunque hasta la fecha no conocemos estudios que demuestren la existencia de un perfil sérico angiogénico en pacientes adultos con malformaciones vasculares, al menos teóricamente sí que puede intuirse, y algunos resultados preliminares no publicados de nuestro grupo de trabajo así lo avalan. Marler et al29 han observado niveles de metaloproteinasas y bFGF en la orina de niños con hemangiomas y malformaciones vasculares en comparación con un grupo control. Otro trabajo demuestra niveles séricos aumentados de bFGF en un paciente con glomangiomas30. Además, algunas observaciones clínicas y estudios moleculares han demostrado que las malformaciones arterio-venosas cerebrales presentan angiogénesis y remodelamiento vascular31. Recientemente hemos apreciado en un paciente con una malformación arterio-venosa extensa y activa unos niveles plasmáticos aumentados de angiopoietina-2 como también ocurre en algunas lesiones cerebrales32 y receptor para Tie-233. Todos estos hallazgos sugieren un papel significativo de la angiogénesis en el desarrollo y mantenimiento de las malformaciones vasculares, quizás no tan abrupto y circunscrito en el tiempo como ocurre con la fase de proliferación de los hemangiomas34,35, pero nada desdeñable de cara a explicar la fisiopatología de las lesiones.
Características clínicas
A continuación seguimos la clasificación de Mulliken y Glowacki5 con los cambios anteriormente comentados respecto a las malformaciones venulares y arterio-venosas12.
Malformaciones venulares
En la clasificación de Mulliken y Glowacki5 se denominan capilares, aunque la histopatología alterada en las vénulas postcapilares del plexo papilar hace que sea más correcto el término de venulares12. Se dividen en malformaciones venulares de la línea media y las clásicamente venulares también denominadas mancha en vino de Oporto, nevus telangiectásico o nevus flammeus.
Las lesiones de la línea media son máculas de coloración rosada, confluentes o no, presentes siempre desde el nacimiento, que aparecen en la línea media de la cabeza y vulgarmente se conocen como «mancha salmón», «mordedura de la cigüeña» o «beso del ángel». Ocurren en el 40 % de los recién nacidos de raza blanca y en el 30 % de los negros. Habitualmente son transitorias y tienden a desaparecer en el primer año de vida en el 65 % de los niños y en el 54 % de las niñas36, sobre todo las de localización anterior. Al contrario que las manchas en vino de Oporto, nunca progresan y su hipertrofia o aspecto en empedrado es extraordinariamente raro. Cuando afectan a la porción anterior se extienden característicamente en forma de V por la glabela y la frente, por el territorio inervado por los nervios supratroclear y supraorbitario. Con relativa frecuencia la nariz, en la zona supra-alar, y el labio superior en los dos tercios del filtrum, se ven afectados.
La mancha en vino de Oporto es una mácula de coloración rosada-rojiza, que con el tiempo va oscureciéndose, y aunque siempre es congénita puede no hacerse aparente hasta varios días después del nacimiento. Ocurre en el 0,4 % de los recién nacidos, sin distinción de sexos. En el 83 % de los casos aparece en la cabeza y el cuello37 (fig. 1) y, curiosamente, afecta más a la hemifacies derecha que a la izquierda38.
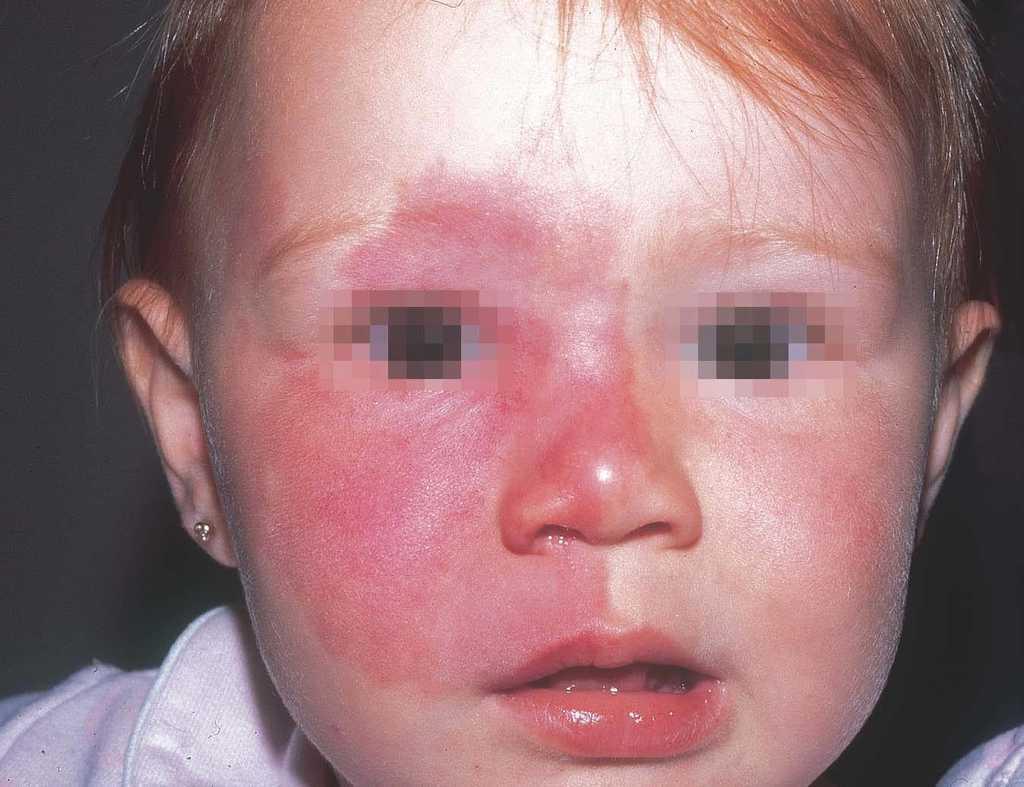
Figura 1. Mancha en vino de Oporto en hemifacies derecha sin sobrepasar la línea media. Afectación de V1 y V2.
Las manchas en vino de Oporto se localizan en uno o más dermatomos faciales definidos por las ramas del nervio trigémino. El dermatomo V2 es el más implicado (57 %), seguido del mandibular (V3) y del oftálmico (V1)37. Cuando hay más de un dermatomo afectado la asociación más frecuente es V2 junto a V1 o a V3 en el 90 % de los casos. La morfología de la lesión generalmente es geográfica confluente, observándose en estos casos mejor respuesta al tratamiento con láser que cuando es parcheada37. La afectación de V2 implica el compromiso de la mucosa adyacente o en continuidad con la lesión cutánea, en concreto del bermellón labial, mucosa labial y mucosa maxilar y gingival. Al contrario que las lesiones de la línea media, estas malformaciones venulares se van oscureciendo y abultando con la edad, adquiriendo un tono más violáceo y un aspecto en empedrado, aunque esta progresión es muy variable individualmente, y es mucho más evidente en el polo cefálico que en otras localizaciones anatómicas (fig. 2). Algunas veces pueden asociarse pequeños cambios esqueléticos en forma de hipertrofia ósea, sobre todo cuando la lesión en V2 se extiende hasta la mucosa gingival y maxilar. Esta hipertrofia localizada favorece una separación de los dientes y un aumento de volumen del labio afecto realmente llamativo, que requerirá corrección quirúrgica (fig. 3).
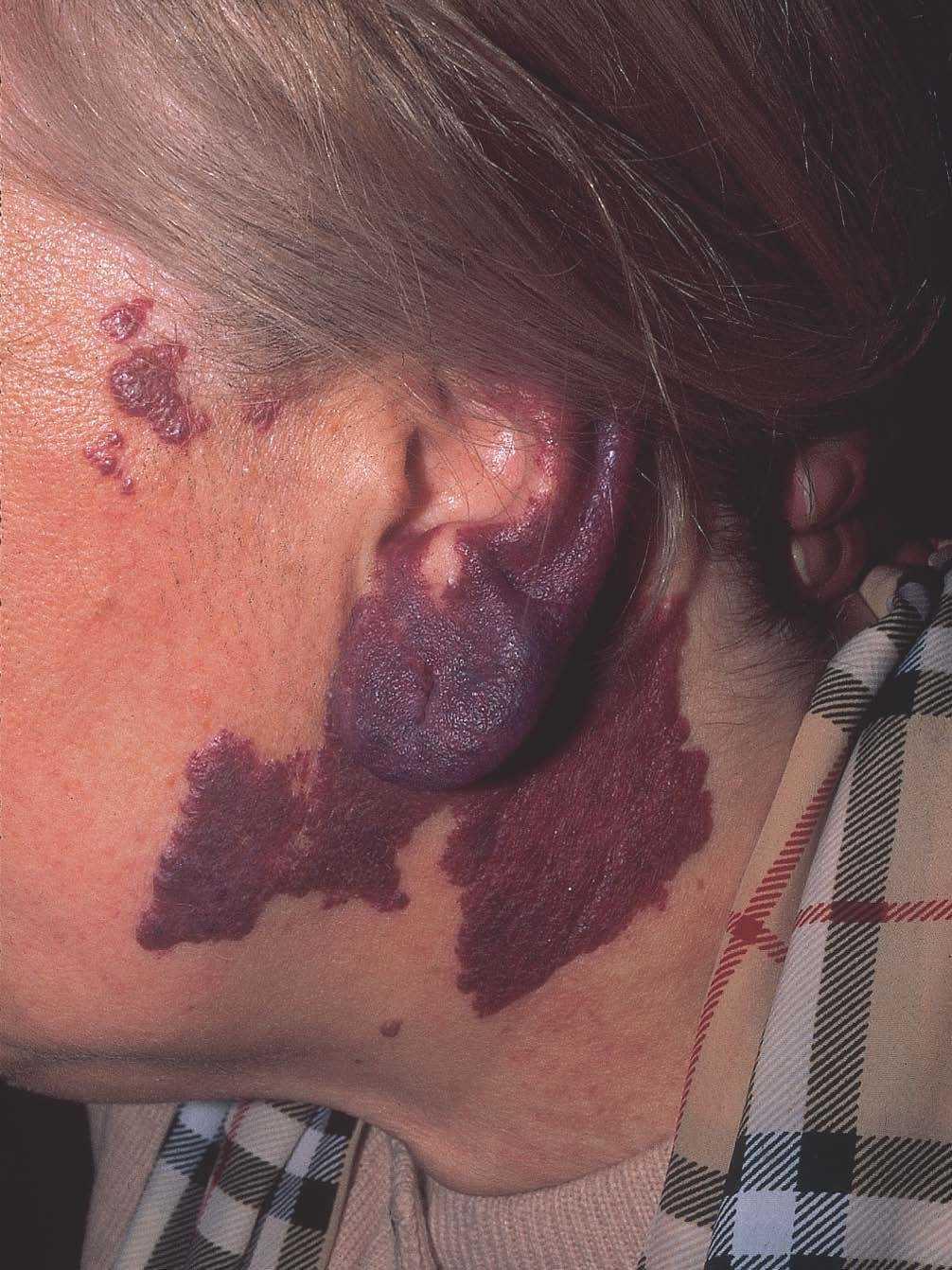
Figura 2. Paciente adulto con mancha en vino de Oporto. Nótese la coloración morada y la hipertrofia del pabellón auricular.
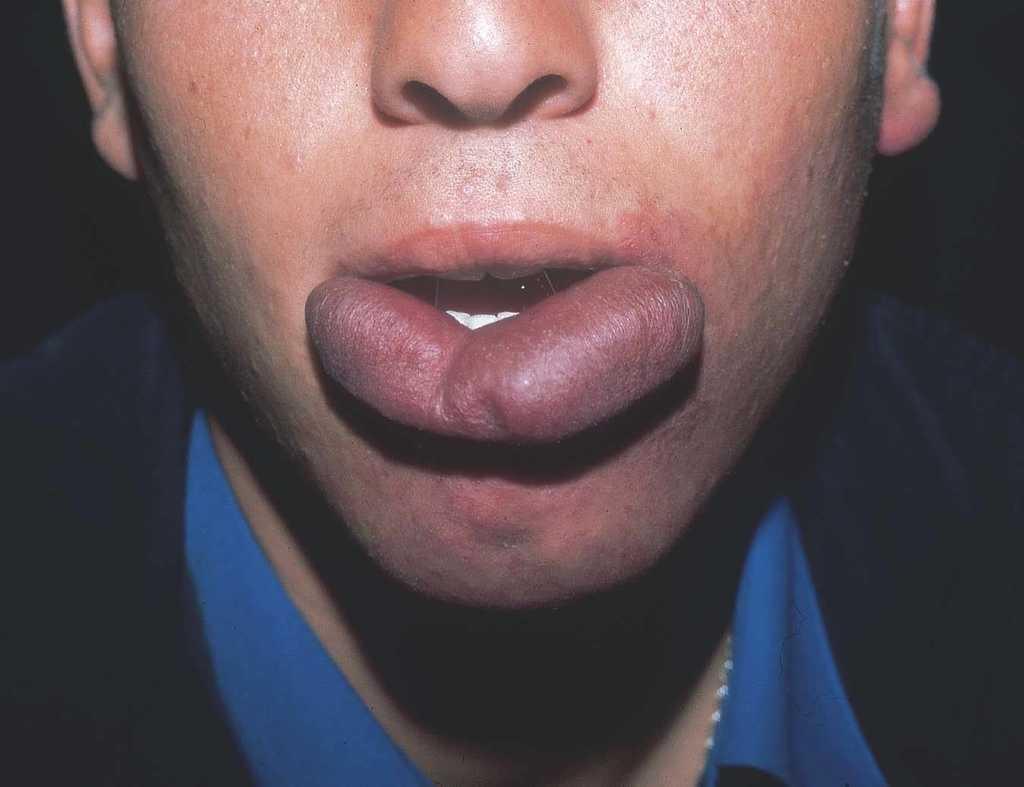
Figura 3. Adolescente con mancha en vino de Oporto de disposición bilateral limitada a V3 con importante hipertrofia labial susceptible de reducción quirúrgica.
Síndromes asociados a malformaciones venulares
Determinadas malformaciones venulares, con una localización anatómica concreta, se asocian a cuadros sindrómicos que son diagnosticados o sospechados por la anomalía vascular cutánea (tabla 5) .

Síndrome de Sturge-Weber o angiomatosis encefalotrigeminal
El síndrome de Sturge-Weber presenta un carácter esporádico, con una incidencia de 1/50.000 neonatos. Clínicamente se caracteriza por una malformación venular facial asociada a una malformación vascular leptomeníngea y a alteraciones oculares39,40, aunque no siempre se observan todas las lesiones, e incluso pueden existir alteraciones leptomeníngeas sin malformación venular. En algunos pacientes se ha demostrado una expresión incrementada del gen de la fibronectina en fibroblastos obtenidos de tejidos lesionados, atribuyéndole un papel en la patogénesis41.
La mancha en vino de Oporto siempre compromete al dermatomo V1. Aunque generalmente es unilateral, puede afectar a ambos lados de la cara o extenderse a la mitad inferior facial y al tronco (fig. 4). Con frecuencia se asocia hiperplasia gingival, labial o de la hemicara afecta a expensas de partes blandas u óseas, en mayor grado cuanto más extensa sea la mancha en vino de Oporto42. La manifestación clínica ocular más importante es el glaucoma, que debe tratarse de forma temprana43. Sin embargo, la manifestación más frecuente es el aumento de vascularización de la coroides que da una imagen característica en el fondo de ojo como de «tomate kétchup». Esta lesión, que suele ser asintomática en la niñez, puede determinar un desprendimiento de la retina en la vida adulta44.
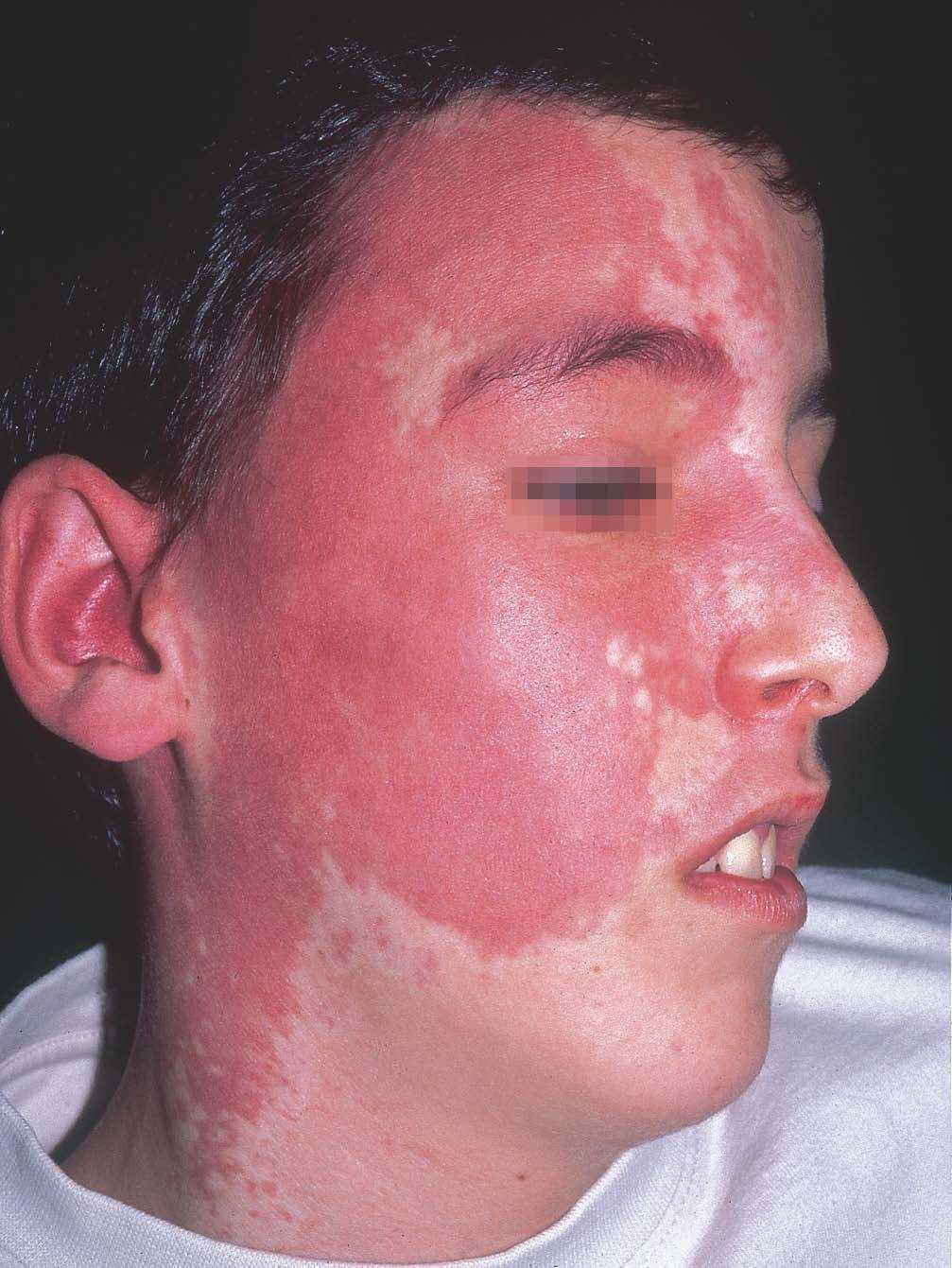
Figura 4. Síndrome de Sturge-Weber con mancha en vino de Oporto a nivel de V1 y V2, glaucoma ocular y afectación leptomeníngea.
La angiomatosis leptomeníngea suele ser ipsilateral a la malformación venular, y clínicamente se manifiesta como un cuadro epiléptico con crisis tónico-clónicas focales en el hemicuerpo contralateral, de aparición durante el primer año de vida43. La respuesta al tratamiento es desigual y la mayoría de las crisis suelen hacerse resistentes a los fármacos antiepilépticos, produciendo una hemiparesia lentamente progresiva. El 50 % de los pacientes presenta retraso mental en la niñez como consecuencia del consumo crónico de antiepilépticos, de la hipoxia local producida por las crisis epilépticas prolongadas y de la atrofia cerebral progresiva45.
Aunque el diagnóstico es esencialmente clínico, la resonancia magnética (RM) permite observar la existencia o no de la alteración vascular leptomeníngea y los signos de atrofia cerebral. Esta técnica también es útil para detectar las llamadas «calcificaciones cerebrales en vía de tren» que con el tiempo aparecen en los pacientes46. Se debe realizar un examen oftalmológico anual que incluya fondo del ojo y presión ocular. En 22 pacientes con síndrome de Sturge Weber (edad entre 8 días y 25 meses) se realizó una tomografía por emisión de fotón único (SPECT) con xenon 133 para ver el flujo sanguíneo regional cerebral, al mismo tiempo que un escáner y una RM. Cuando había convulsiones ya establecidas la SPECT demostró una disminución del flujo, confirmándose una hipoperfusión e hipometabolización del lado dañado. Sorprendentemente, cuando la SPECT se hizo en pacientes antes de tener las convulsiones, el lado afecto mostraba hiperperfusión en el 75 % de los casos. Por tanto, esta prueba puede diagnosticar precozmente un daño cerebral en el síndrome de Sturge-Weber, con su repercusión terapéutica, ya que tratar las convulsiones precozmente limita la anoxia y el daño neurológico47.
Síndrome de Cobb o angiomatosis cutáneo-meningoespinal
Asocia una malformación venular de distribución metamérica en tronco o porción proximal de extremidades, suprayacente a una malformación vascular de la médula espinal. La malformación espinal comienza a dar síntomas en la niñez o en la adolescencia en forma de paraplejía o paraparesia espástica y pérdida sensitiva por debajo del nivel medular afecto. Ante una mancha en vino de Oporto metamérica en tronco hay que sospechar este síndrome antes de que aparezcan las manifestaciones neurológicas, ya que en algunos pacientes la lesión medular es tributaria de tratamiento quirúrgico. La confirmación diagnóstica se realiza mediante RM48.
Aunque en menor medida a como ocurre con los hemangiomas, la presencia de una malformación venular en la región lumbosacra puede asociarse a un disrafismo espinal, que habrá que descartar en todos los pacientes mediante una prueba de imagen (ecografía o RM).
Cutis marmorata telangiectásica congénita
Aunque este cuadro se incluye dentro de las malformaciones venulares simples, se trata en realidad de una malformación mixta que combina elementos venulares y venosos, con una incidencia estimada de 1/3.000 neonatos y un patrón de herencia autosómico recesivo49,50. Clínicamente se caracteriza por un eritema reticulado de patrón liveloide, que asemeja un aspecto marmóreo, con lesiones planas o deprimidas y telangiectasias en su superficie. En caso de enfermedad segmentaria, puede existir hipotrofia o atrofia de la piel de la extremidad afecta o más raramente ulceración. En algunos pacientes con el tiempo las lesiones tienden a aclararse progresivamente hasta desaparecer por completo51. Cuando las lesiones son difusas, siguen un patrón en mosaico, o existe afectación cefálica, se recomienda la realización de exploraciones complementarias para descartar otras patologías extra-cutáneas asociadas, como alteraciones retinianas, glaucoma neovascular secundario, persistencia del ductus arterioso o espina bífida52.
Facomatosis pigmentovasculares
Se trata de procesos congénitos caracterizados por la combinación de nevus vasculares y melanocíticos en un mismo paciente, representando una manifestación de mosaicismo en «manchas gemelas». La mayoría de los pacientes pueden clasificarse en 4 tipos. El tipo 1 asocia una malformación venular con nevus pigmentocelular y nevus epidérmico verrucoso. En el tipo 2 se observa la malformación venular asociada a melanocitosis dérmica tipo mancha mongólica aberrante. En el tipo 3 se combinan malformación venular y nevus spilus. El tipo 4 es la asociación de malformación venular, nevus spilus y mancha mongólica aberrante. Estos tres últimos pueden presentar también un nevus anémico. Últimamente ha sido descrito el tipo 5, en el que se aprecia cutis marmorata telangiectásica congénita extensa y mancha mongólica aberrante53. A su vez, cada tipo se subdivide en A y B según exista afectación exclusivamente cutánea (A), o presente alguna repercusión sistémica (oftalmológica, del sistema nervioso central o anomalías esqueléticas) o relación con los síndromes de Sturge-Weber, Kippel-Trenaunay y/o nevus de Ota (B)54.
Malformaciones linfáticas
Clásicamente han recibido diferentes nombres: linfangioma, higroma quístico, linfangioma circunscrito y linfangiomatosis55. Como el resto de las malformaciones vasculares, las linfáticas siempre son congénitas, aunque sólo el 65-75 % son diagnosticadas en el nacimiento, cifra que alcanza el 80-90 % al final del segundo año de vida56. La localización más frecuente es la cabeza y sobre todo el cuello (90 %)57, propugnando algunos autores la complejidad del sistema linfático cervical para explicar esta elevada presentación58. Otras localizaciones típicas son axila, tórax, mediastino, retroperitoneo, nalgas y región anogenital. El aspecto clínico varía en función del tamaño, profundidad y localización de la lesión, siendo frecuente la presencia de múltiples vesículas traslúcidas, de contenido viscoso a nivel cutáneo o mucoso, cuyo aspecto tiene un parecido a los «huevos de rana» (fig. 5). La piel circundante es normal, a veces con una tonalidad azulada. Las lesiones superficiales están conectadas a cisternas linfáticas más profundas en tejido subcutáneo o submucoso. En función de la profundidad, las malformaciones se dividen en variante microquística o difusa, también conocida como linfangioma, caracterizada por bordes mal definidos y edema masivo generalizado, y variante macroquística o localizada, compuesta por quistes multitabicados. En general, las lesiones mucosas (que afectan al suelo de la boca, las mucosas yugales y la lengua) son del primer tipo y resultan difíciles de erradicar con cirugía, mientras que las lesiones cervicales, las más frecuentes, también conocidas como higromas quísticos, son macroquísticas y más fáciles de extirpar quirúrgicamente. Éstas últimas se presentan como masas indoloras, de consistencia gomosa, no pulsátiles, generalmente recubiertas por piel de color normal.
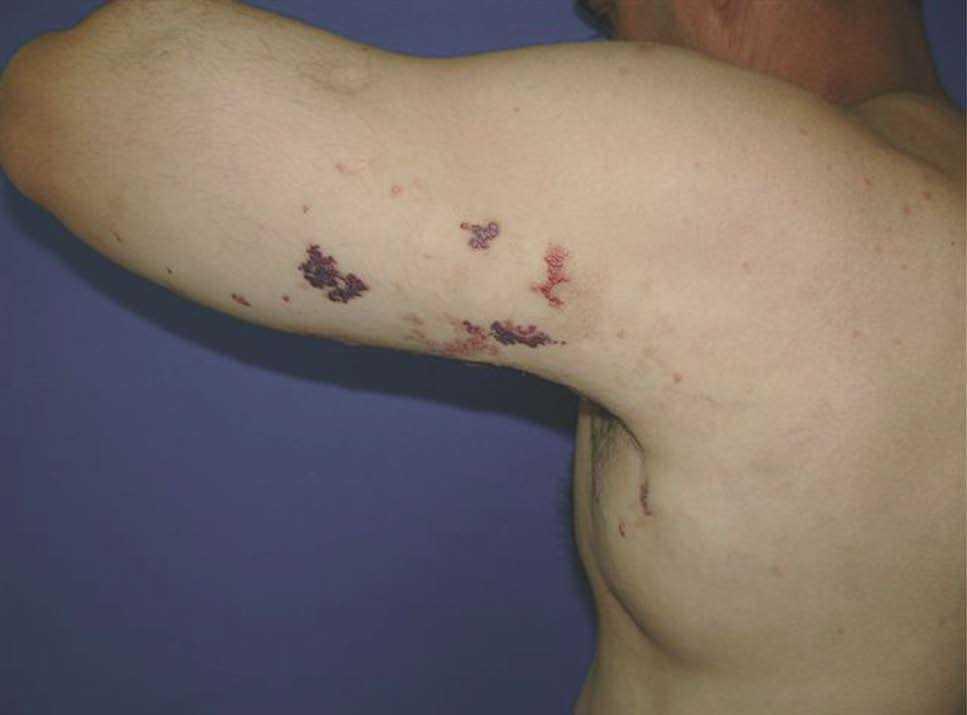
Figura 5. Malformación linfática. Nótese la lesión macroquística axilar abultada y vesículas hemorrágicas en la superficie flexora proximal del antebrazo.
Con relativa frecuencia las lesiones microquísticas de mucosa pueden exacerbarse coincidiendo con un traumatismo accidental o terapéutico (quirúrgico), una hemorragia intralesional o una infección59. Por extensión, las lesiones cervicales pueden comprimir la faringe o la tráquea cuando hay afectación mediastínica, y las cercanas a la órbita producir proptosis. El crecimiento brusco de una malformación linfática cervical puede ser una emergencia al comprometer la vía aérea, asociando en ocasiones distrés respiratorio. Algunas series reportan episodios de sepsis hasta en un 16 % de las lesiones cervicales60, posiblemente desencadenada por la flora bacteriana oral10. Las lesiones profundas producen hipertrofia esquelética en el 83 % de los afectados, con distorsión evidente en el 33 % de ellos61, lo cual no se explica como en otras malformaciones vasculares por un incremento en el aporte sanguíneo. Concretamente, la hipertrofia mandibular puede ocasionar prognatismo y mala oclusión. Las anomalías de los linfáticos torácicos, sobre todo del conducto torácico, pueden comenzar con sintomatología pleural en forma de quilotórax, y las del tracto gastrointestinal causar hipoalbuminemia debido a la pérdida proteica por enteropatía. Las lesiones pélvicas pueden manifestarse por obstrucción urinaria, diarrea e infecciones recurrentes. Cuando afectan a una extremidad, las grandes malformaciones linfáticas con frecuencia producen dolor, inflamación y gigantismo a expensas del crecimiento de tejido músculo-esquelético. En las malformaciones combinadas tipo síndrome de Klippel-Trenaunay, cuanto mayor es la extensión linfática, mayor es el número de complicaciones y efectos secundarios.
Existe un síndrome denominado Gorham-Stout, también conocido como «enfermedad ósea fantasma», caracterizado por la presencia de malformaciones linfáticas y venosas de localización cutánea, mediastínica y ósea. Las lesiones óseas suelen ser unilaterales y provocan osteolisis con fibrosis secundaria, pudiendo ocasionar, si son extensas, la desaparición de huesos enteros62,63.
Se han descrito algunos casos de regresión de malformaciones vasculares linfáticas, teóricamente explicables por la aparición con el tiempo de una derivación linfático-venosa64, por lo que algunos aconsejan retrasar el tratamiento10,58,59, aunque posiblemente algunas de esas lesiones «involucionadas» correspondan erróneamente a hemangiomas subcutáneos que siguen su evolución natural. Hoy en día, mediante el uso de marcadores selectivos de células linfáticas (LYVE-1, D-240, etc.), es posible detectar la implicación de vasos linfáticos en algunas malformaciones vasculares que a priori no son especialmente linfáticas. Algunos autores postulan que la presencia de estas células se asocia a un mayor número de trastornos de la coagulación que más adelante se comentan65.
Malformaciones venosas
Las malformaciones venosas están formadas por vasos ectásicos, de bajo flujo sanguíneo, morfológica e histológicamente similares a las venas. Se dividen en superficiales o profundas, localizadas, multicéntricas o difusas. La piel o mucosa que las cubre varía de color en función de la profundidad y el grado de ectasia de la lesión. Las más superficiales son de color morado, y las profundas más azuladas, verdosas o incluso imperceptibles. Se trata de lesiones blandas al tacto, a veces de aspecto nodular, que se vacían con la compresión (fig. 6). En determinadas posiciones, la malformación se rellena de sangre, como por ejemplo las localizadas en cabeza y cuello tras realizar una maniobra de Valsalva, y en otras se vacía cuando se eleva la zona afecta por encima del nivel del corazón. En las lesiones muy ectásicas no es rara la presencia de pequeñas trombosis venosas que pueden sobreinfectarse y expandirse rápidamente con dolor e inflamación, y de flebolitos que son marcadores radiológicos de este tipo de malformaciones y aparecen a edades tempranas. También son especialmente característicos el dolor matutino que va disminuyendo con el movimiento y la exacerbación sintomática en las mujeres con el embarazo y los cambios hormonales10 (fig. 7).
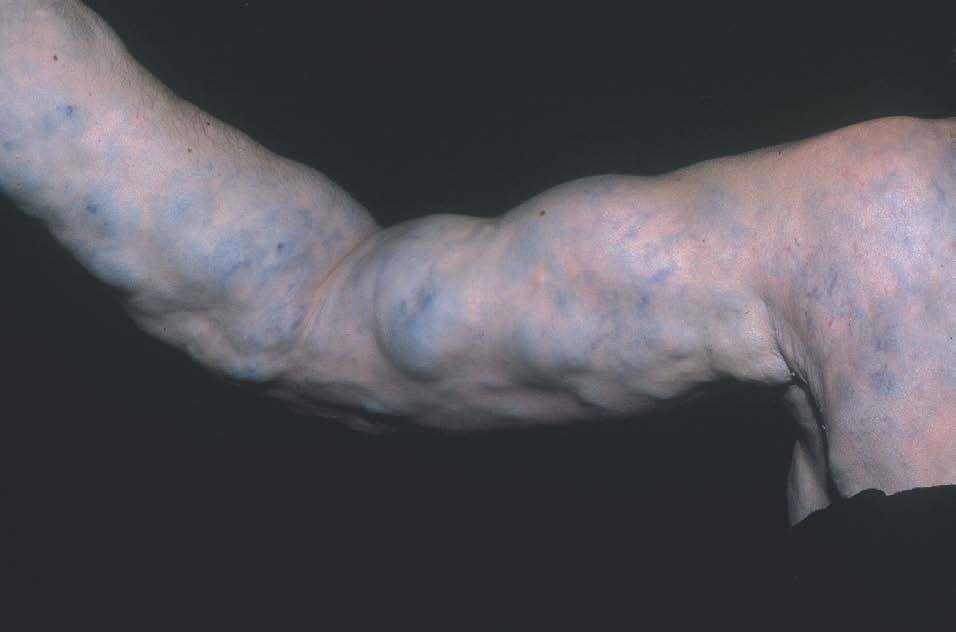
Figura 6. Malformación venosa extensa en extremidad superior derecha. Lesiones depresibles que se vacían parcialmente al elevar el miembro.
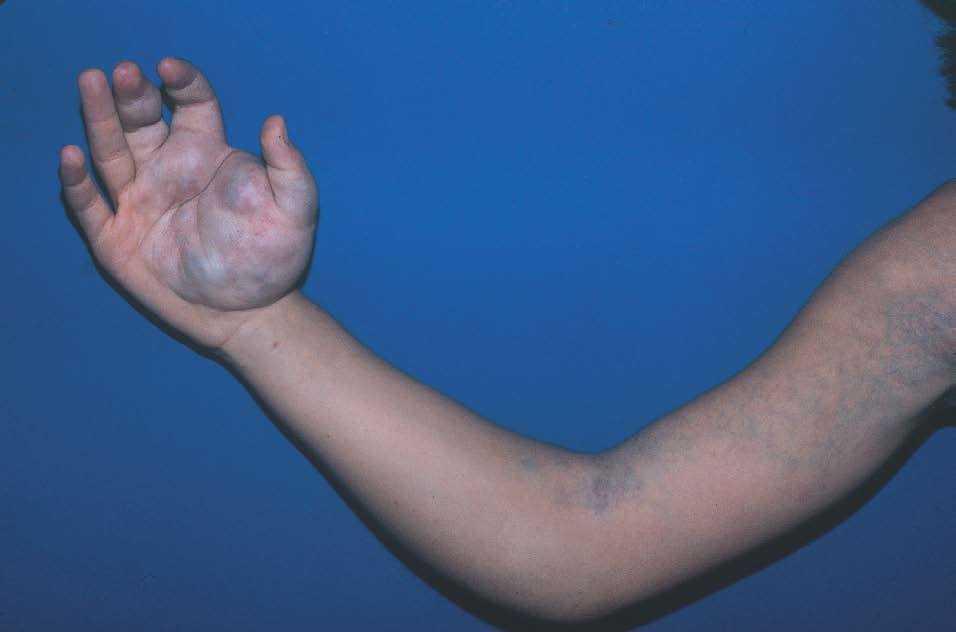
Figura 7. Malformación venosa bien definida en la mano que se extiende difusamente por la extremidad superior derecha.
En el polo cefálico las malformaciones venosas con frecuencia presentan compromiso mucoso, afectan la lengua, el paladar, los labios y las mucosas yugales, e infiltran músculo, glándulas salivares e incluso estructuras óseas (figs. 8 y 9). Concretamente la afectación mandibular o maxilar no es rara, tratándose de masas indoloras de lento crecimiento. Lo más llamativo es la separación dental, con el movimiento o caída precoz de alguna pieza con hemorragia intensa asociada. Radiológicamente, el hueso afecto se presenta como una imagen en panal de abejas66. La hipertrofia de tejidos blandos y hueso perilesional es relativamente frecuente en lesiones de gran tamaño. Cuando afectan a la región periorbitaria o al cuello, el crecimiento puede ocasionar compromiso ocular y obstrucción de la vía aérea respectivamente. Con relativa frecuencia las malformaciones venosas asientan en los miembros, muchas veces con una extensión lineal y profunda mayor que la apariencia externa. Prácticamente siempre hay afectación muscular, y no es raro el compromiso articular y óseo. Al contrario que en las malformaciones vasculares combinadas tipo síndrome de Klippel-Trenaunay, no suele haber hipertrofia músculo-esquelética de la extremidad afecta, sino más bien atrofia o hipotrofia (fig. 10). Cuando está interesada la rodilla es frecuente la gonalgia con limitación funcional y hemartrosis, y no es raro que la artropatía por hemosiderina acabe evolucionando a una artritis degenerativa. En una serie de 176 pacientes con malformaciones venosas que afectaban tejido músculo esquelético, se apreció una doble incidencia en mujeres, siendo dos tercios diagnosticadas en el nacimiento y el resto en la infancia o adolescencia. La localización más frecuente fue la cabeza, el cuello y las extremidades, y el dolor y la inflamación los síntomas principales67. En otra serie de 27 pacientes con malformaciones venosas extensas de miembros, todos presentaban afectación muscular, el 81 % afectación articular (codo o rodilla) y el 63 % de 19 pacientes estudiados, alteraciones óseas (adelgazamiento, desmineralización o cambios líticos)68. Otro grupo demuestra desmineralización ósea local en el 71 % de los pacientes, que puede asociarse a fracturas patológicas61. Las grandes malformaciones venosas de miembros obligan a descartar una extensión al tronco y una afectación visceral, concretamente las de miembros superiores a pleura, mediastino y pulmón, y las de miembros inferiores a pelvis y cavidad abdominal. Por este motivo se postula la realización de una prueba de imagen completa (angio-TC o angio-RM) para estadiar la extensión.

Figura 8. Gran malformación venosa en hemifacies con expansión hacia el cuello en mujer joven. Se aprecian cicatrices post-quirúrgicas en esta última localización.
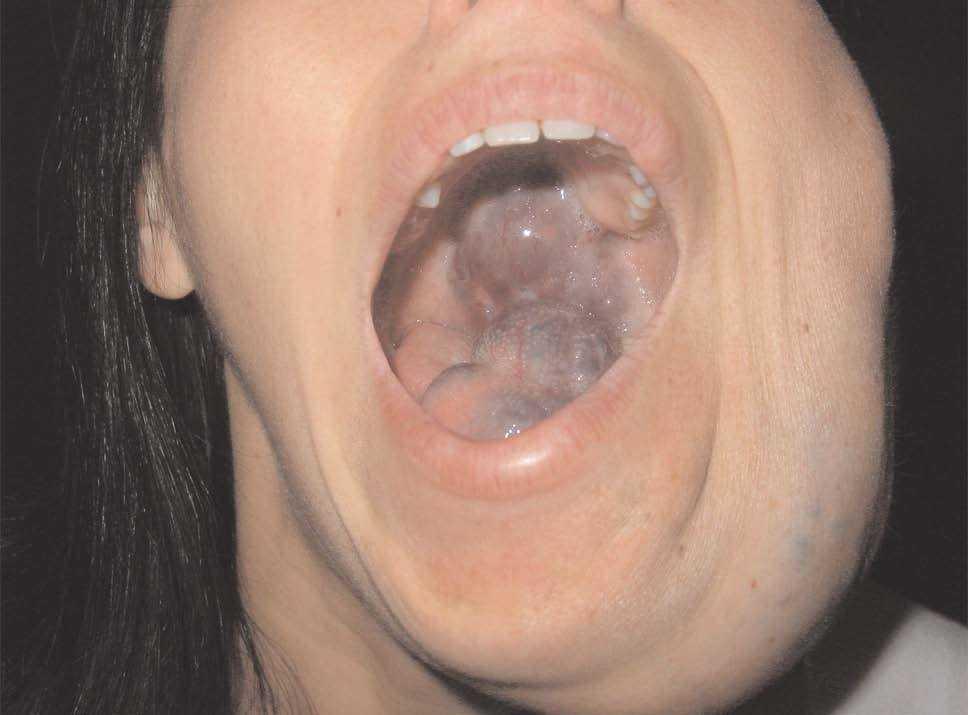
Figura 9. Detalle de la paciente anterior con afectación extensa de paladar y mucosa oral.
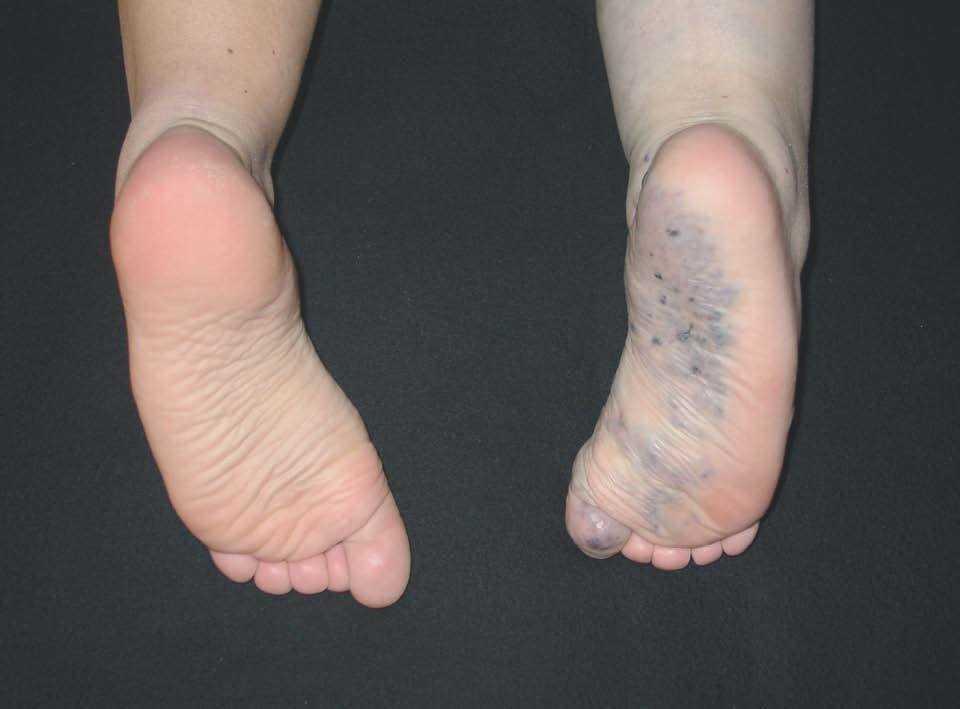
Figura 10. Malformación venosa en miembro inferior con afectación plantar. Destaca atrofia ósea y músculo-esquelética contrariamente a lo que suele ocurrir en malformaciones combinadas tipo síndrome de Klippel-Trenaunay.
En este grupo de malformaciones venosas puras de patrón segmentario o en malformaciones combinadas veno-linfáticas extensas (tipo Klippel-Trenaunay, que más adelante se comenta), se han descrito trastornos de la coagulación. Concretamente una coagulación intravascular localizada, diferente de la coagulación intravascular diseminada del síndrome de Kasabach-Merrit. Es importante incidir aquí en que este síndrome, caracterizado por anemia hemolítica, trombocitopenia y coagulopatía, es específico de algunos tumores vasculares como el hemangioendotelioma kaposiforme o el hemangioma en penacho, pero no de los hemangiomas comunes ni de las malformaciones vasculares independientemente del tipo69. Recientemente se ha descrito una nueva entidad denominada linfangioendoteliomatosis multifocal con trombocitopenia, caracterizada por lesiones vasculares congénitas cutáneas y gastrointestinales que se encuentran a caballo entre un tumor y una malformación vascular, y por alteraciones de la coagulación, también a caballo entre una coagulación intravascular diseminada tipo síndrome de Kasabach-Merrit y una coagulación intravascular localizada70. Curiosamente tanto en esta nueva entidad como en los dos tumores descritos, existen unos vasos linfáticos anómalos que además de LYVE-1 son positivos para VEGF-R3, una tirosín cinasa expresada por células endoteliales linfáticas71. Así podría postularse que estas alteraciones de la coagulación con atrapamiento plaquetario aparecen especialmente en tumores o malformaciones vasculares con diferenciación linfática. En la coagulación intravascular localizada, característica de algunas malformaciones vasculares, se produce un consumo local de factores de la coagulación dentro de la malformación o secundario al éstasis venoso, como consecuencia del cual se forman los microtrombos y los flebolitos. En estos pacientes el fibrinógeno suele estar bajo (< 0,5 g/l), y el dímero-D elevado. La determinación basal de estos parámetros es útil de cara a la realización de un procedimiento terapéutico, ya que la retirada de las medias de compresión, la cirugía, o la escleroterapia pueden empeorar el cuadro predisponiendo a un posible tromboembolismo, por lo que debe iniciarse anticoagulación con heparina de bajo peso molecular.
Finalmente, en pacientes con malformaciones venosas extensas de extremidades y en el síndrome de Klippel-Trenaunay, pueden encontrarse anomalías del sistema venoso profundo en el 47 % de los casos, concretamente flebectasias (36 %), aplasia o hipoplasia de troncos venosos (8 %), aneurismas (8 %) y avalvulia (7 %)16. Estos hallazgos obligan a realizar un estudio del sistema venoso profundo del miembro afecto, sobre todo antes de realizar un procedimiento terapéutico por las posibles complicaciones que puedan derivarse.
Cuadros con entidad propia
Tumor glómico. Glomangiomas
Son tumores raros que tienen su origen en las células de músculo liso localizadas en derivaciones arterio-venosas de partes acras. Aunque la localización más frecuente es la cutánea o subcutánea, también se han descrito fuera de la piel en hueso, estómago, colon, tráquea y mediastino72. Cuando son múltiples se denominan glomangiomas, que algunas veces tienen carácter hereditario73, y deben diferenciarse del síndrome del nevus azul en tetina de goma (tabla 5).
La evolución natural y las manifestaciones clínicas facilitan la distinción entre malformaciones venosas y glomuvenosas. Las últimas son lesiones nodulares o en forma de placa, rosadas o azul oscuras con una apariencia en empedrado y a veces áreas hiperqueratósicas, sobre todo aquellas localizadas de forma segmentaria en una extremidad (fig. 11). Generalmente afectan a la piel y al tejido celular subcutáneo sin invadir planos profundos, y son dolorosas a la palpación74. Por el contrario, las malformaciones venosas son de un color más azul verdoso, depresibles, invaden con facilidad el músculo y las articulaciones, presentan flebolitos y son dolorosas, principalmente por la mañana, empeorando en relación con cambios hormonales. Estos pacientes pueden presentar cuadros de coagulación intravascular localizada, que no han sido apreciados en las malformaciones glomuvenosas. En caso de duda, la presencia de células glómicas en el estudio patológico es concluyente en el diagnóstico75.
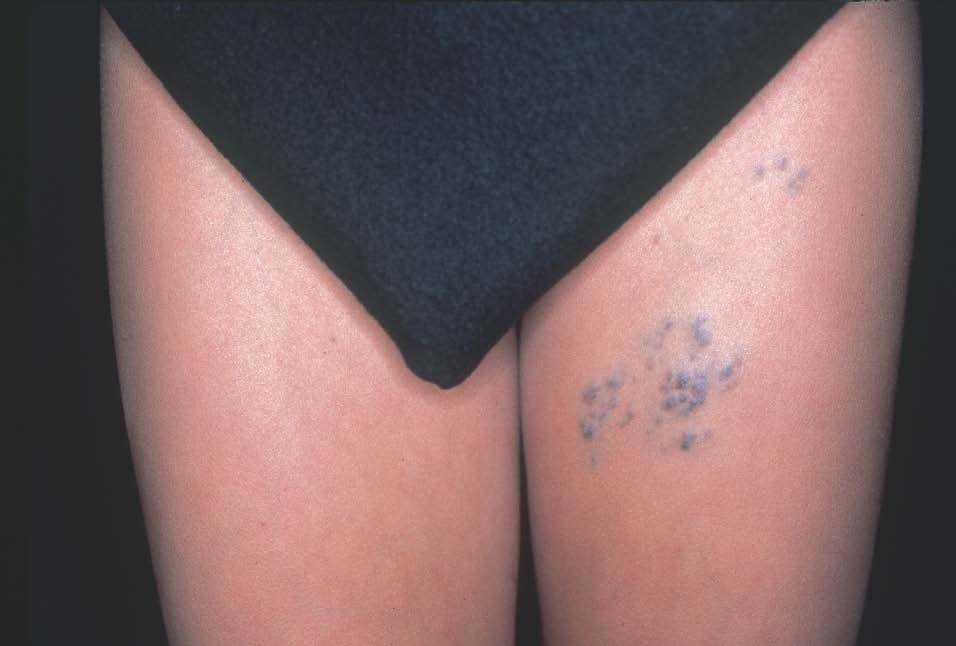
Figura 11. Varón de 16 años con malformación glomuvenosa tipo placa siguiendo patrón dermatómico en muslo.
Síndrome del nevus azul en tetina de goma76,77
Es una entidad que asocia múltiples malformaciones venosas en la piel y el tracto gastrointestinal. Las lesiones cutáneas, que generalmente están presentes en el momento del nacimiento o bien aparecen de manera progresiva en la primera infancia, son nódulos de pequeño tamaño, azulados o purpúricos. En ocasiones se trata de tumoraciones más extensas o máculas azuladas. Característicamente son depresibles a la palpación en forma de tetina de goma, y es típico que provoquen dolor de manera espontánea. En el tubo digestivo la localización principal de las lesiones es el intestino delgado. Estas malformaciones pueden ser causa de hemorragias digestivas con anemia secundaria y requerirán control endoscópico y tratamiento quirúrgico en función de su gravedad. Desde el punto de vista histopatológico, se trata de ectasias vasculares de forma y tamaño irregular, localizadas en la dermis profunda y en el tejido celular subcutáneo. La RM es útil en el diagnóstico de la extensión visceral y se emplea para el estudio de familiares asintomáticos. La mayoría de los casos publicados son esporádicos, aunque en algunos se ha descrito un patrón de herencia autosómico dominante78.
Malformación venosa cutáneo-mucosa familiar
Se debe a una mutación en el receptor Tie-2, que se hereda con un patrón autosómico dominante17. Clínicamente se caracteriza por la presencia de múltiples lesiones venosas de pequeño tamaño, que pueden afectar la piel y las mucosas18.
Malformaciones venosas cerebrales (también denominadas «angiomas cavernosos»)
Presentan un carácter familiar con el mismo patrón hereditario que las anteriores. Un subgrupo de estos pacientes tienen lesiones cutáneas capilares-venosas de aspecto hiperqueratósico79, y con frecuencia pueden asociar una malformación venosa ocular. En algunas familias afectas se ha detectado una anomalía en 7q11-2280.
Malformaciones arterio-venosas
Las malformaciones arterio-venosas hacen referencia a un grupo de malformaciones congénitas formadas por varios trayectos fistulosos que comunican arterias con venas. En la literatura a veces se recogen con otros sinónimos como: mancha de nacimiento pulsátil, aneurisma cirsoide o aneurisma arterio-venoso. Mulliken y Young12 prefieren reservar el título de fístula arterio-venosa para la variante traumática adquirida, compuesta por una única fístula. El análisis histológico de las malformaciones arterio-venosas en niños y recién nacidos revela que el nidus de la lesión está compuesto de capilares. Según la malformación va madurando, el grado de ectasia aumenta y el desarrollo de la dilatación venosa e hipertrofia arterial se hacen aparentes.
La localización más frecuente de las malformaciones arterio-venosas es la intracraneal, afectándose luego por orden de frecuencia la cabeza, el cuello, los miembros, el tronco y las vísceras. Dentro de las malformaciones vasculares, las arterio-venosas a pesar de ser congénitas igual que el resto constituyen el grupo que más tardíamente se diagnostica, a veces durante la cuarta-quinta década de la vida10. Erróneamente pueden confundirse con un hemangioma o más con una mancha en vino de Oporto (fig. 12), pero las malformaciones arterio-venosas suelen ser máculas levemente sobreelevadas, más calientes y a veces con latido palpable. También son frecuentes la ulceración, el dolor intenso, el sangrado intermitente y la hipertrofia ósea subyacente a la lesión. Una malformación arterio-venosa proximal con un gran flujo sanguíneo puede aumentar el gasto y originar un fallo cardiaco congestivo81, aunque lo normal es que el gasto cardiaco aumentado esté compensado durante años15,82; por el contrario, si la malformación es distal hay una propensión a reducir el flujo y producir isquemia periférica (síndrome del robo) (fig. 13). La historia natural de una malformación arterio-venosa está documentada en el estadiaje clínico descrito por Schobinger, expuesto más arriba. Al contrario que las malformaciones venosas, estas lesiones no se vacían totalmente tras la compresión, se rellenan con rapidez y son más firmes a la palpación. De forma similar a como ocurre con otras malformaciones su extirpación parcial, manipulación, traumatismo y cambios hormonales pueden favorecer su crecimiento. Dentro del grupo de las malformaciones vasculares, las arterio-venosas son las más activas, con mayor posibilidad de expansión y crecimiento y, al mismo tiempo, más difíciles de tratar83. Generalmente su extensión histológica supera con creces la apariencia clínica, apreciándose infiltración microscópica del tejido subyacente que favorece la recidiva tras una extirpación parcial. Además en ellas se ha demostrado un patrón proangiogénico, ya comentado, que favorece el crecimiento y la activación.
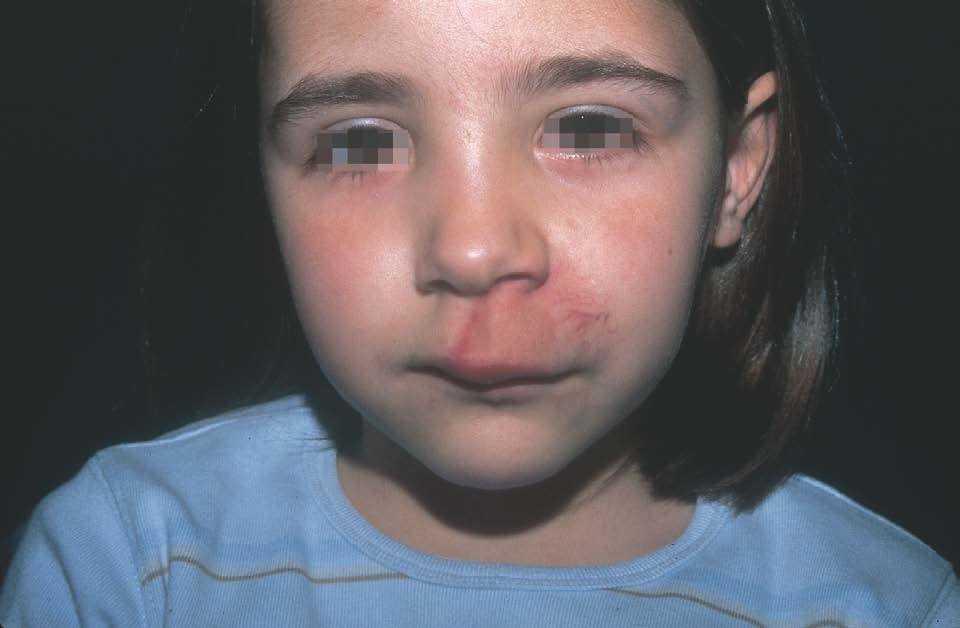
Figura 12. Niña de 6 años afecta de mancha en vino de Oporto en labio superior asociada a hipertrofia. Tras estudio angiográfico se realizó confirmación diagnóstica de malformación arterio-venosa.

Figura 13. Malformación arterio-venosa en mano con síndrome de robo y necrosis distal, con amputación del primer dedo e importantes alteraciones tróficas en segundo dedo.
Malformaciones mixtas o combinadas
Constituyen complejos sindrómicos que casi siempre se asocian a sobrecrecimiento de tejido músculo-esquelético (tabla 5). Se pueden dividir según el flujo sanguíneo sea bajo o alto.
De bajo flujo
Síndrome de Klippel-Trenaunay
Se caracteriza por la asociación de una malformación vascular venular, linfática y venosa, junto con hipertrofia esquelética y aumento de tejidos blandos de uno o más miembros10,84. Aunque la etiopatogenia permanece desconocida, Servelle85 postula que la obstrucción o atresia del sistema venoso profundo produce una hipertensión venosa crónica que va a ser la responsable de la mancha en vino de Oporto, las venas varicosas y la hipertrofia de la extremidad86. Aunque la presentación es de aparición esporádica, recientemente se han descrito casos familiares con mutaciones genéticas22,87,88. La malformación venular o mancha en vino de Oporto suele ser múltiple, afecta generalmente una extremidad inferior (95 % de los casos) y con frecuencia se extiende a los glúteos y al tórax. En más de un 10 % de los pacientes la lesión sobrepasa el miembro y aparece afectación de tronco e incluso hemicorporal. Una publicación reciente distingue entre manchas de bordes «geográficos» (contorno irregular, bien definido, asemejando a un continente) de coloración rojiza intensa o violácea (figs. 14 y 15), de otras más «difusas», con una distribución segmentaria, de coloración rosada (fig. 16). Las primeras presentan más afectación linfática y conllevan un mayor número de complicaciones89. Las nuevas técnicas diagnósticas abren nuevas perspectivas en pacientes con síndrome de Klippel-Trenaunay. Los avances en las angiografías no invasivas como la venografía por RM en 3D y/o la venografía por TC multicorte permiten una evaluación completa de estos pacientes con una única prueba de imagen. Con estos procedimientos, en una serie pequeña pero significativa, hemos apreciado cómo las manchas geográficas también se asocian con más frecuencia a hipoplasia o atresia del sistema venoso profundo90.
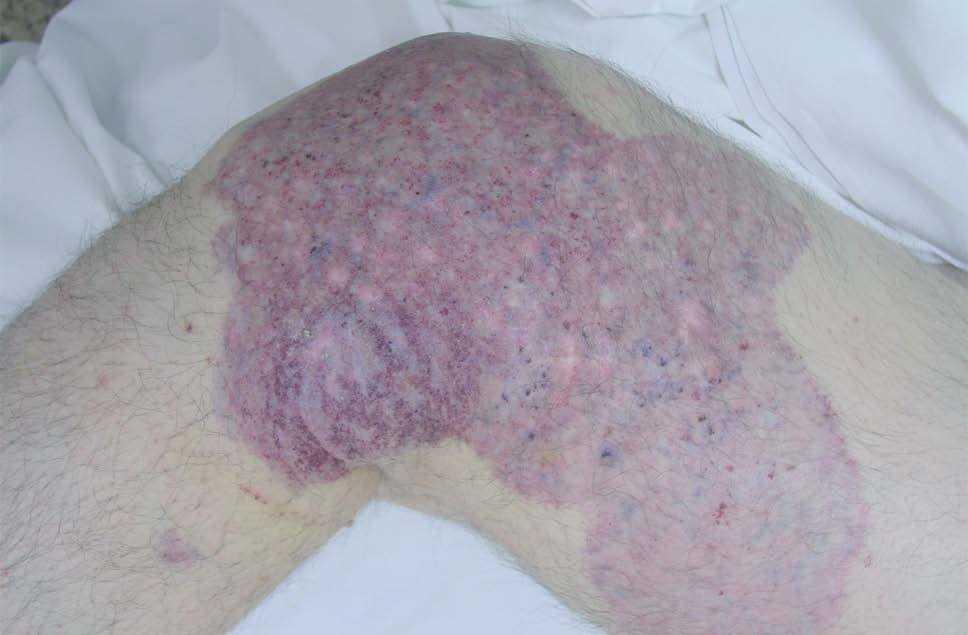
Figura 14. Mancha en vino de Oporto geográfica en rodilla en paciente con síndrome de Klippel-Trenaunay. Se aprecia color violáceo y bordes bien definidos.
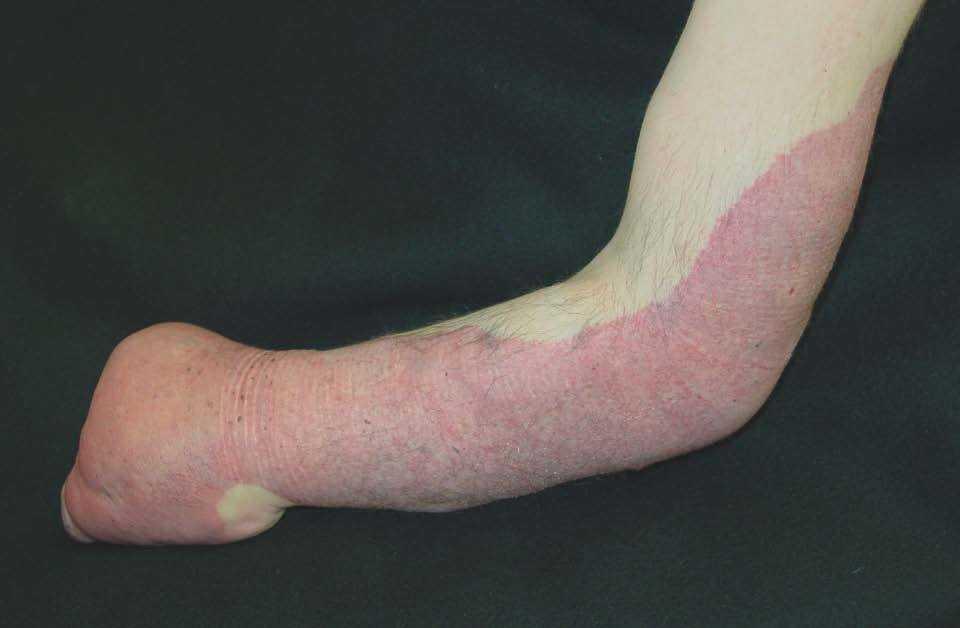
Figura 15. Mancha en vino de Oporto geográfica en brazo en paciente con síndrome de Klippel-Trenaunay hemicorporal. Presenta asociada una deformidad distal de la extremidad afecta.
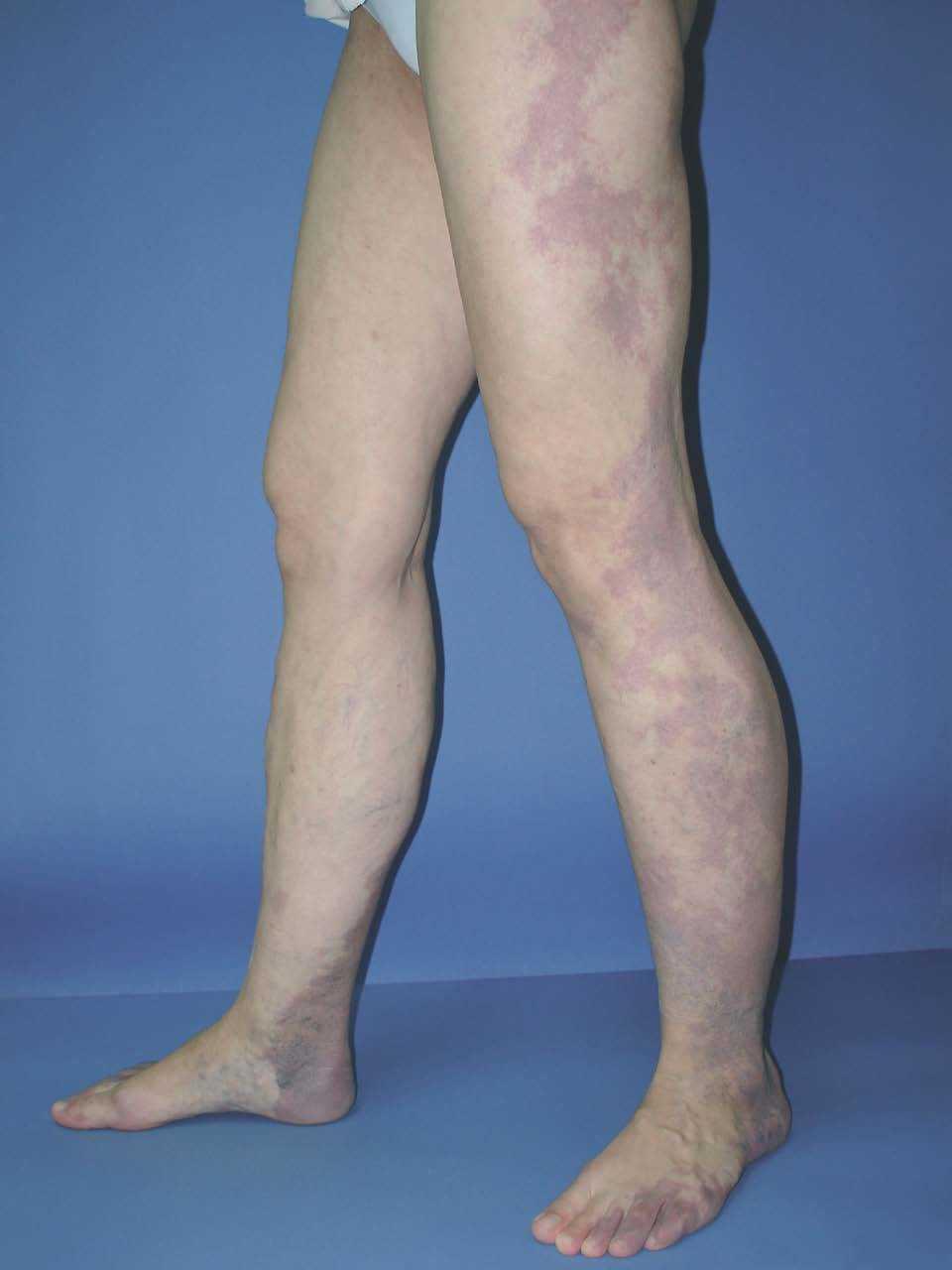
Figura 16. Mancha en vino de Oporto difusa de bordes y coloración tenues en la cara externa de extremidad inferior en un paciente con síndrome de Klippel-Trenaunay.
La malformación venosa se manifiesta como venas laterales anómalas o venas embrionarias persistentes que son prominentes por incompetencia valvular y por la alteración frecuentemente asociada del sistema venoso profundo91. Aunque se pueden observar desde el nacimiento, aparecen con más frecuencia cuando el paciente inicia la deambulación. Hay hiperplasia linfática en más del 50 % de los pacientes, generalmente en forma de vesículas de contenido claro o hemorrágico en la superficie cutánea, que se asocia a linfedema y a macroquistes linfáticos (figs. 17 y 18).

Figura 17. Malformación veno-linfática de afectación genital. Vesículas negruzcas (hemorrágicas) características sobre mancha geográfica violácea.
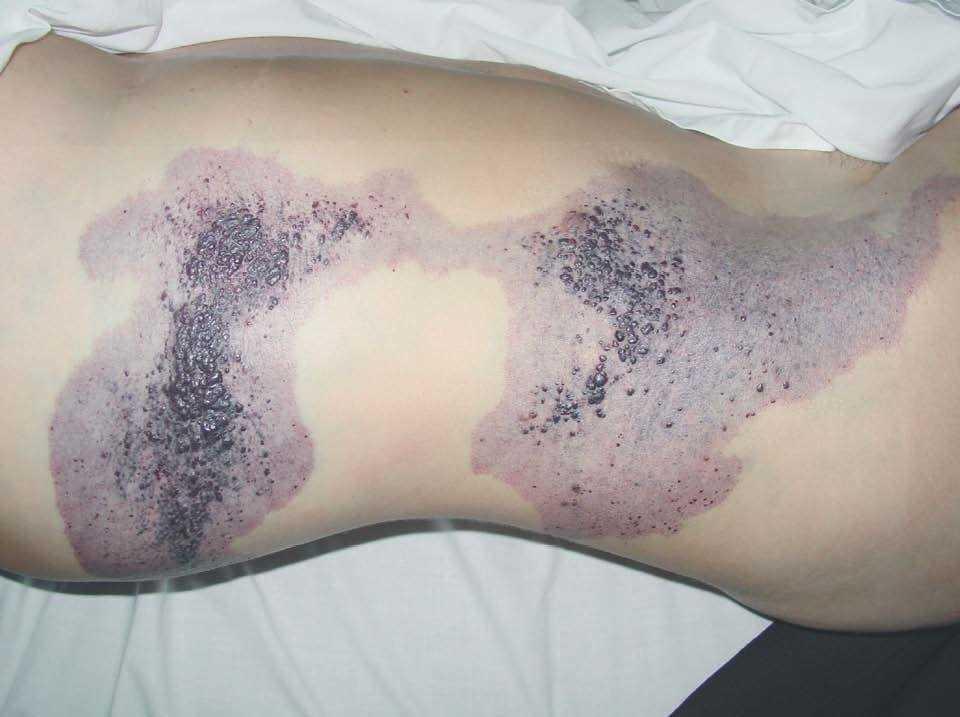
Figura 18. Mujer afecta de malformación veno-linfática extensa, con mancha geográfica en tórax-abdomen y lesiones similares que se extienden por la extremidad inferior.
El aumento de la extremidad afecta va haciéndose llamativo con la edad destacando un aumento de tejidos blandos y macrodactilia que requerirán seguimiento y tratamiento específicos, aunque en un pequeño porcentaje de pacientes hay atrofia y acortamiento de la extremidad92. La principal complicación del síndrome de Klippel-Trenaunay es la tromboflebitis, que ocurre en el 20-45 % de los pacientes93,94 y ocasiona embolismos pulmonares en el 4-25 % de los casos95; también son frecuentes una coagulopatía en forma de coagulación intravascular localizada, un hemotórax y sangrado intestinal o urinario por compromiso focal. A mayor afectación linfática mayor riesgo de celulitis de repetición y bacteriemias96.
Síndrome de Proteus
Es un cuadro heterogéneo definido por la presencia de lesiones vasculares, esqueléticas y de tejidos blandos, de disposición asimétrica y extensión variable97. Pueden observarse lesiones cutáneas lineales verrucosas, lipomas y lipomatosis, macrocefalia, miembros asimétricos con gigantismo parcial de mano, pie o ambos y un engrosamiento cerebriforme plantar característico que histológicamente corresponde a un colagenoma. En una serie de 55 pacientes 98,2 % presentaba crecimiento corporal asimétrico y macrodactilia98. Las lesiones vasculares ocurren en el 69 % de los casos y son, por orden de frecuencia, malformaciones venulares tipo mancha en vino de Oporto (fig. 19), malformaciones linfáticas (micro y macroquísticas) y malformaciones combinadas de bajo flujo tipo síndrome de Klippel-Trenaunay. Nunca se han descrito lesiones de alto flujo como las del síndrome de Parkes Weber.
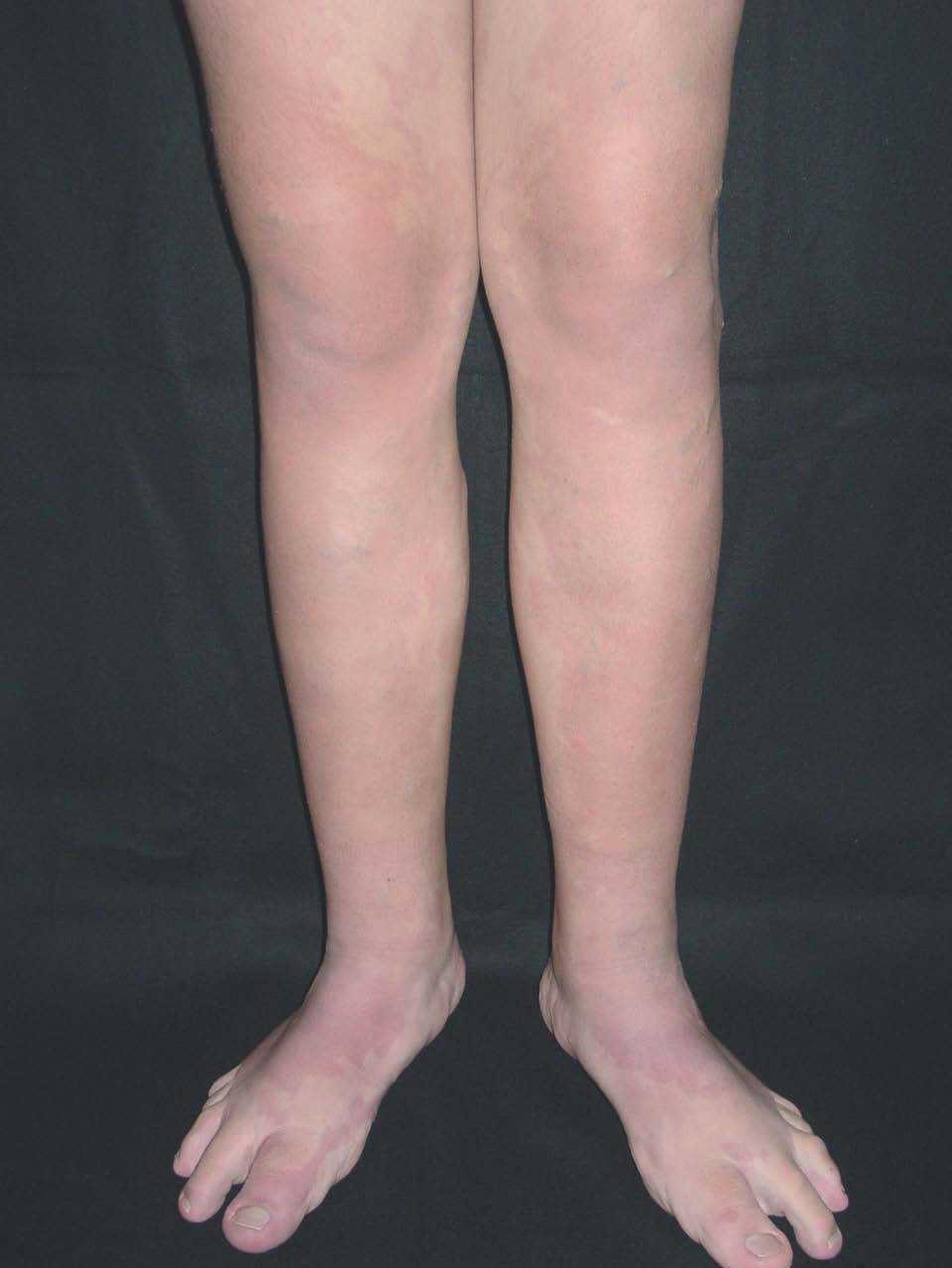
Figura 19. Varón de 17 años afecto de síndrome de Proteus. Mancha en vino de Oporto bilateral e hipertrofia de pies con macrodactilia.
Síndrome de Maffucci
Es una displasia mesenquimal congénita y esporádica, que asocia malformaciones venosas, capilares y con menos frecuencia linfáticas, con exóstosis y encondromas99-101. Ocasionalmente pueden aparecer hemangioendoteliomas de células fusiformes en las lesiones vasculares preexistentes. Las lesiones pueden ser localizadas habitualmente en manos y pies o generalizadas y estar presentes al nacer, aunque lo más frecuente es un desarrollo precoz y progresivo de múltiples nódulos azulados subcutáneos de consistencia blanda, que a menudo contienen flebolitos.
De manera simultánea se desarrollan encondromas, caracterizados clínicamente como nódulos duros, en los huesos largos sobre todo en las manos o en los pies, que causan deformidades más o menos evidentes y fracturas patológicas en función del grado de afectación102. Los encondromas radiológicamente presentan un aspecto hipodenso, y su principal complicación es el riesgo de degeneración maligna, sobre todo a condrosarcomas, cuya alta incidencia alrededor del 40 % hace obligatoria la realización de una biopsia de cualquier lesión dolorosa.
De alto flujo
Síndrome de Parkes Weber
Se trata de una malformación venular arterio-venosa. Aparece en el nacimiento y afecta más al miembro inferior (77 %) aunque en menor proporción que el síndrome de Klippel-Trenaunay. Se caracteriza por una mácula rosado-rojiza difusa de bordes geométricos o difusos que va aumentando de forma simétrica. A diferencia del síndrome de Klippel-Trenaunay, la lesión vascular es de alto flujo con fístulas arterio-venosas, no suele presentar venas laterales anómalas, las malformaciones linfáticas y la afectación músculo-esquelética son raras y ocurre en los brazos hasta en un 23 % de los casos103,104. En lugar de tromboflebitis y riesgo de embolismo pulmonar, la principal complicación en el síndrome de Parkes Weber es el aumento del gasto que puede originar fallo cardiaco e isquemia cutánea12,105.
Conflicto de intereses
Declaro no tener ningún conflicto de intereses.
Correspondencia:
Pedro Redondo.
Departamento de Dermatología.
Clínica Universitaria de Navarra. 31080 Pamplona. España.
predondo@unav.es
Aceptado el 24 de enero de 2007.


