


Six classic variants of porokeratosis (PK) have been described, only 3 of which (disseminated superficial PK [DSP], disseminated superficial actinic PK, and PK palmaris, plantaris et disseminata) are disseminated forms, characterized by a gradual onset and a mainly asymptomatic course.1,2
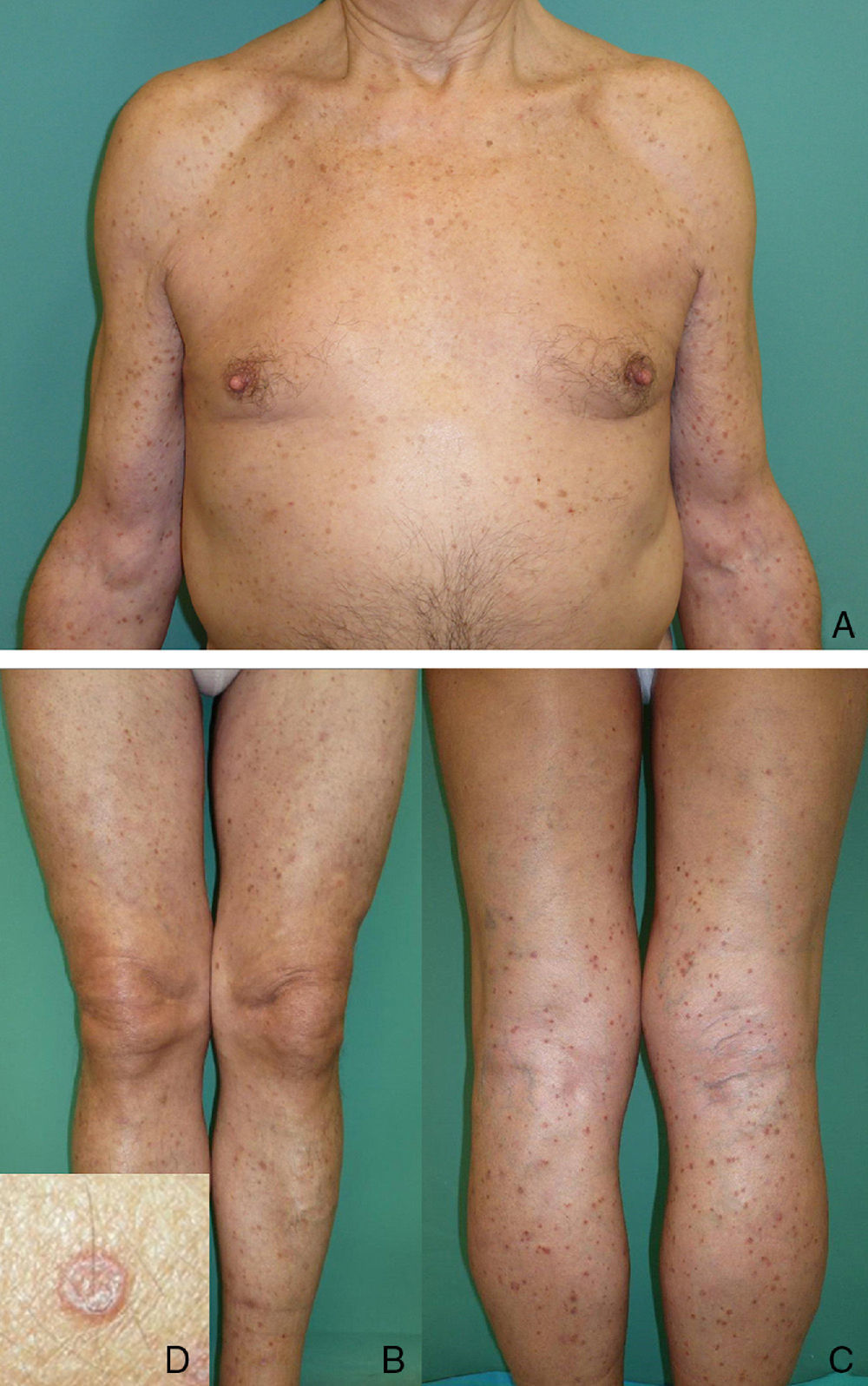
We present the case of a white man of 79 years of age with no past medical history of interest. He attended our clinic for the first time in February 2007 for multiple erythematous-brownish, slightly keratotic, and intensely pruritic papules that had arisen 2-and-a-half years earlier. The lesions had presented a rapid onset on the forearms and lower third of the legs, spreading to the limbs and trunk (Fig. 1); the number of lesions and the inflammatory activity had been variable. Successive skin biopsies revealed the presence of narrow columns of parakeratotic cells that crossed the stratum corneum, associated with an absence or reduction of the underlying stratum granulosum, marked spongiosis, and a dense superficial perivascular inflammatory infiltrate formed of lymphocytes and occasional eosinophils (Fig. 2). In view of the intense pruritus, successive or combined treatments were tried with various topical agents, narrowband UV-B phototherapy, oral prednisone at different doses, acitretin, oral antihistamines, and serotonin reuptake inhibitors, none of which produced any significant improvement. In June 2011 the patient started treatment with oral ciclosporin, 4mg/kg/d, which led to almost complete remission of his symptoms. Dose reductions below 1mg/kg/d or treatment interruption were always followed by an increase in the number and inflammatory activity of the lesions and an unrelenting pruritus very poorly tolerated by the patient. Treatment with ciclosporin has therefore been maintained at doses varying between 1 and 4mg/kg/d, looking to achieve the minimal effective dose, with brief periods of treatment interruption, enabling the patient to maintain an adequate quality of life. No adverse effects have arisen to date, and blood tests and the blood pressure have remained within normal limits.
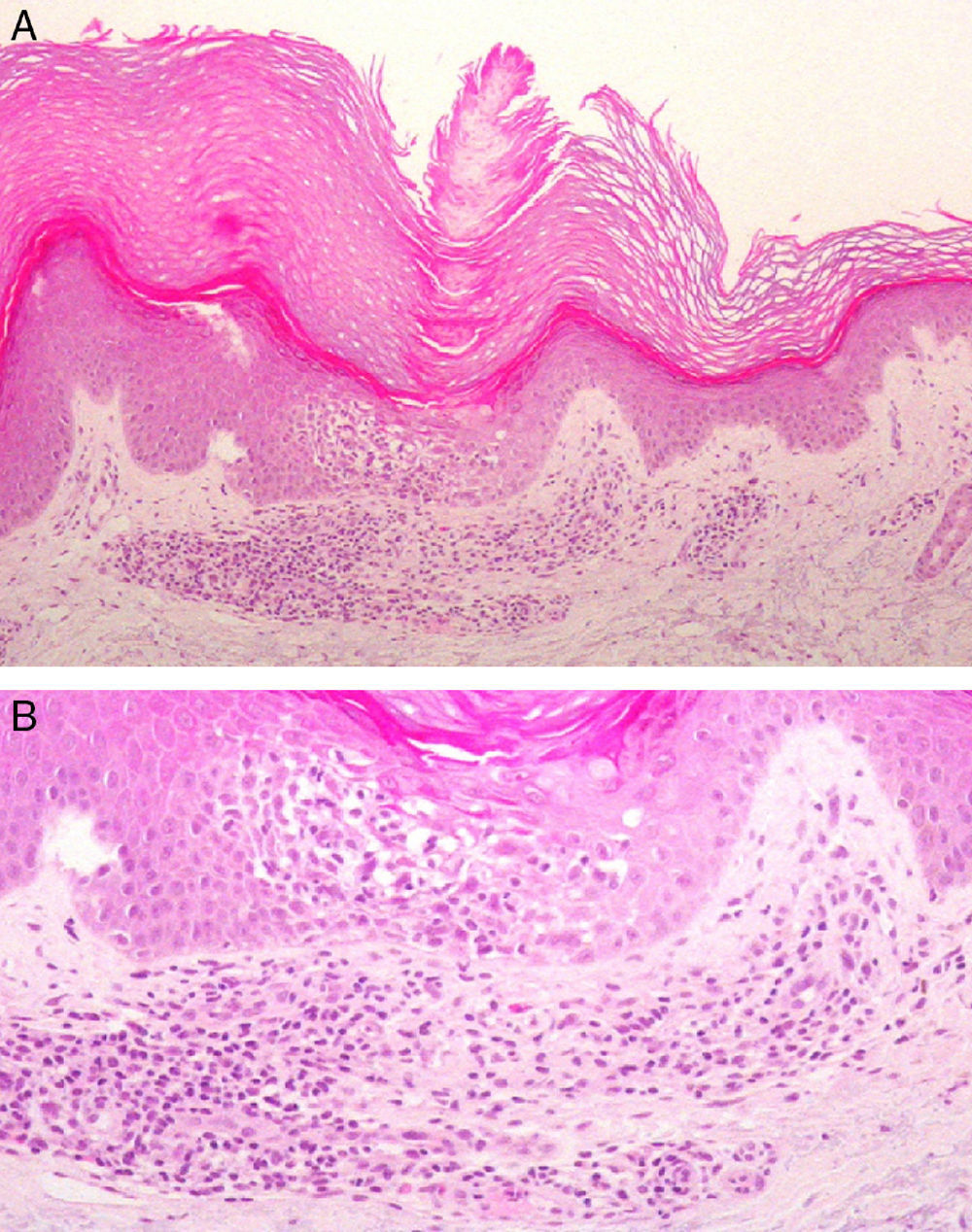
A, Cornoid lamella with intense underlying spongiosis and a dense superficial perivascular inflammatory infiltrate. Hematoxylin and eosin, original magnification ×100. B, Spongiosis and lymphocyte exocytosis in the epidermis beneath the cornoid lamella. There is an inflammatory infiltrate formed mainly of lymphocytes, with occasional eosinophils. Hematoxylin and eosin, original magnification ×200.
In 1992, Kanzaki et al.3 coined the term eruptive pruritic papular PK to refer to a previously undescribed condition of intensely pruritic monomorphic lesions of rapid onset in 3 Asian patients with a long-standing history of asymptomatic DSP. In 1995, a fourth case, identical to the previous ones, was published under the name of inflammatory disseminated superficial PK, after observing spongiosis and a dense lymphocytic inflammatory infiltrate on skin biopsy, supporting the hypothesis of an immune reaction against clones of mutated keratinocytes.4 The first case in a white patient was reported in 1997.5 That patient had no history of PK lesions and required systemic treatment to control the disease, contrasting with the spontaneous resolution observed in previous cases. A more recent publication described the case of an Asian man with a 13-year history of frequent periods of exacerbation.6 As in our case, no underlying disease was identified that could explain the persistent activity.
Recently, in order to reduce the conceptual confusion surrounding this rare variant, Shoimer et al.7 proposed unification of all the previous terminology under the name eruptive disseminated PK (EDP), to include all forms of rapid onset (less than 2 months), with multiple lesions and a histology compatible with PK. Additionally, they found that approximately 60% of cases were part of a paraneoplastic syndrome or were associated with immunosuppression. In our opinion, the term EDP is perhaps not the most appropriate. First, the mean clinical course of many previously published cases exceeds the proposed 2-month limit or was not even specified. And second, it should be recognized that the intense pruritus, present in almost all cases and frequently refractory to treatment,3,5,6,8 is one of the condition's most notable traits. We therefore consider that the previously used terms “pruritic” and “inflammatory”, are more suitable, especially when the histopathology findings of this variant are also taken into account.9
With the exception of a limited number of cases that have required a period of systemic treatment,5 the final tendency of the disease is towards spontaneous resolution within a few months to a year.3,4,8,10 No cases of malignant degeneration have been reported to date, though this has been observed in up to 10% of other variants of PK.1 The typically limited clinical course of this disease would probably explain the absence of an association with malignant tumors. The persistent disease activity after 7 years of follow-up in our patient is of particular note, as is the good response to ciclosporin, a therapeutic option not previously described in the literature.
We therefore propose the term “inflammatory disseminated pruritic porokeratosis” for this variant, and we have presented a case with frequent periods of exacerbation, unrelenting pruritus, and a good response to ciclosporin, drawing attention to the use of this therapeutic option in resistant cases.
Please cite this article as: Montes-Torres A, Camarero-Mulas C, de Argila D, Gordillo C, Daudén E. Poroqueratosis pruriginosa diseminada inflamatoria. Buena respuesta a ciclosporina. Actas Dermosifiliogr. 2016;107:261–262.


