


La infiltración cutánea por células linfoplasmocitarias en la macroglobulinemia de Waldeström (MW) es un hecho muy inusual durante la evolución de esta enfermedad1.
Un varón de 73 años fue diagnosticado en 2011 de MW tras realizar un estudio de anemia y monocitosis casual en un análisis de sangre rutinario. Al diagnóstico, presentaba una afectación masiva de médula ósea, con infiltración por células linfoplasmocitarias de más del 45% del total, con restricción de cadenas ligeras lamba/kappa y componente monoclonal IgM en sangre de 3,84g/dL (normal <2,7g/dL). Además, se realizó el estudio de la mutación L265P en el exón 5 del gen MYD88 (3p22.2) que resultó positivo.
El estudio de extensión mostró adenomegalias axilares, retroperitoneales, ilíacas e inguinales, además de hepatoesplenomegalia. El paciente no refería síntomas de hiperviscosidad y la biopsia de tejido adiposo para amiloide resultó negativa, por lo que se decidió adoptar una actitud expectante.
En septiembre de 2014, consultó en dermatología por la aparición de unas placas eritematosas, ligeramente descamativas y asintomáticas en zona superior de tronco, espalda y cara (fig. 1a). El estudio histológico mostró un infiltrado linfoplasmocitario sugestivo de linfoma B (fig. 2a y b), con positividad predominante de CD20 (relación con CD3, 2:1) y negatividad para CD38 y CD138 (fig 2c y 2d). Las escasas células plasmáticas acompañantes no mostraron restricción de cadenas ligeras lambda/kappa. El estudio genético mostró reordenamiento positivo monoclonal IgH (región FR1) y biclonal IgH (región FR3), por lo que el diagnóstico fue concluyente con infiltración cutánea por un linfoma linfoplasmocítico/MW. No se realizó tinción inmunohistoquímica para IgM en la biopsia cutánea por no estar disponible en el centro.
 Placas eritematodescamativas en tórax y flancos. Presentación inicial. b) Curación de las lesiones cutáneas al mes de finalizar el tratamiento quimioterápico.")
 H-E x2. Infiltrados linfoplasmocitarios en plexo perivascular superficial y profundo. b) H-E x40. Infiltrados linfoplasmocitarios periglandulares. c) CD20×10. Positividad para CD20 de infiltrado linfoplasmocitario. d) CD38×10. Negatividad del infiltrado neoplásico para CD38. Escasas células plasmáticas; no monoclonales; positivas para CD38.")
a) H-E x2. Infiltrados linfoplasmocitarios en plexo perivascular superficial y profundo. b) H-E x40. Infiltrados linfoplasmocitarios periglandulares. c) CD20×10. Positividad para CD20 de infiltrado linfoplasmocitario. d) CD38×10. Negatividad del infiltrado neoplásico para CD38. Escasas células plasmáticas; no monoclonales; positivas para CD38.
Durante el año 2015 se objetivó progresión clínica (astenia, artralgias, pérdida de peso, sudoración), anemización progresiva (hemoglobina <10g/dL) y aumento en el tamaño de las adenopatías previas, por lo que se decidió iniciar tratamiento sistémico con 6 ciclos de rituximab y bendamustina a dosis habituales. Además, se asoció al tratamiento metilprednisolona tópica para las lesiones cutáneas. Un mes después del fin de la quimioterapia las lesiones cutáneas desaparecieron. El paciente refería mejoría de su estado general y se objetivó mejoría analítica (hemoglobina >12g/dL) y radiológica. Actualmente el paciente continúa asintomático desde el punto de general y cutáneo, en seguimiento por el Servicio de Hematología (fig. 1b).
La MW es un trastorno linfoproliferativo de tipo B poco frecuente, de etiología desconocida, caracterizado por proliferación de linfoplasmocitos en la médula ósea y un pico monoclonal de IgM en sangre periférica2.
La afectación cutánea por MW se da en el 5% de los casos de esta enfermedad. Según la clasificación de Libow et al. se diferencian 2tipos de lesiones cutáneas1,3: «neoplásicas» o como consecuencia de infiltración cutánea directa de las células linfoplasmocitarias y «no neoplásicas» o secundarias a la paraproteinemia. Las «no neoplásicas» son las más frecuentes y, a su vez, se clasifican en 3subtipos: aquellas relacionadas con el síndrome de hiperviscosidad (púrpura acral, sangrado de mucosas, edema periférico), las asociadas a crioglobulinemia (cianosis acral, fenómeno de Raynaud, hipersensibilidad al frío, livedo reticularis, vasculitis leucocitoclástica) y las relacionadas con las paraproteínas específicas (dermatitis ampollosa IgM, macroglobulinemia cutis y pápulas eritematosas asociadas a MW).
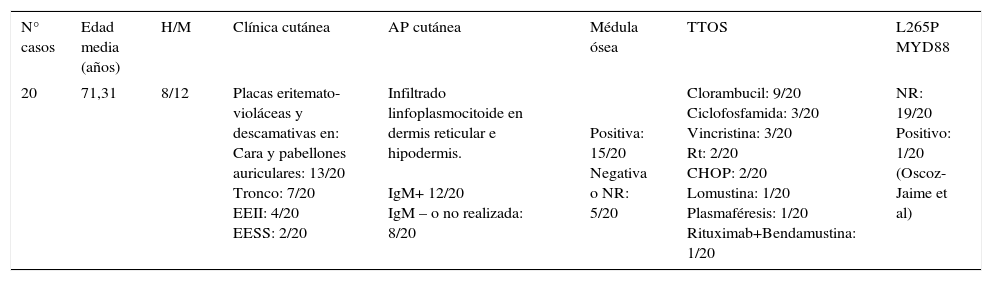
Las lesiones cutáneas «neoplásicas» o como consecuencia de la infiltración por células linfoplasmocitarias son las menos frecuentes. Existen descritos en la literatura únicamente una veintena de casos (tabla 1)3-8 y su diagnóstico en la práctica diaria es excepcional. Se caracterizan por presentarse clínicamente en forma de placas eritematosas, levemente descamativas, algo infiltradas y localizadas de manera preferente en cara y orejas de manera simétrica y en tórax, flancos y espalda3-8. Puede aparecer de manera precoz durante la enfermedad, pero cabe destacar que su presencia no empeora el pronóstico. Las células halladas en las lesiones cutáneas pertenecen a la estirpe linfoplasmocitaria y se caracterizan por presentar positividad para marcadores B (CD19, CD20, CD22) y negatividad para CD38 y CD138 (marcadores de células plasmáticas).
Resumen de casos reportados de afectación cutánea por MW (ver material adicional)
| N° casos | Edad media (años) | H/M | Clínica cutánea | AP cutánea | Médula ósea | TTOS | L265P MYD88 |
|---|---|---|---|---|---|---|---|
| 20 | 71,31 | 8/12 | Placas eritemato-violáceas y descamativas en: Cara y pabellones auriculares: 13/20 Tronco: 7/20 EEII: 4/20 EESS: 2/20 | Infiltrado linfoplasmocitoide en dermis reticular e hipodermis. IgM+ 12/20 IgM – o no realizada: 8/20 | Positiva: 15/20 Negativa o NR: 5/20 | Clorambucil: 9/20 Ciclofosfamida: 3/20 Vincristina: 3/20 Rt: 2/20 CHOP: 2/20 Lomustina: 1/20 Plasmaféresis: 1/20 Rituximab+Bendamustina: 1/20 | NR: 19/20 Positivo: 1/20 (Oscoz-Jaime et al) |
Sexo: H: hombre; M: mujer; EEII: extremidades inferiores; EESS: extremidades superiores. AP cutánea=Anatomía Patológica cutánea.
IgM en AP cutánea medida por inmunofluorescencia directa (IFD): +(positiva), −(negativa).
Biopsia de médula ósea [NR: No registrado, Positivo=compatible con linfoma linfoplasmocítico].
TTOS=Tratamientos realizados. Rt=radioterapia. CHOP=ciclofosfamida+doxorrubicina+vincristina+prednisona.
L265P/MYD88=Mutación L265P en el gen MYD88. NR=no registrado/no realizado.
La diferenciación entre los casos de linfoma linfoplasmocítico/MW y linfoma de la zona marginal (LZM) con intensa diferenciación plasmocítica puede ser compleja en algunos casos, debido a que existen numerosos casos en los que puede existir una superposición clínico-patológica. Se puede recurrir al estudio de la ciclina D1 (positiva en el LZM y negativa en la afectación cutánea de MW) y el estudio de la mutación del gen MYD88 (mutación somática L265P positiva en la mayoría de los casos de MW9) y la traslocación t11;18 (presente en los casos de LZM) para realizar el diagnóstico definitivo de estas entidades10.
Las lesiones análogas a las lesiones «neoplásicas» de la MW en el mieloma múltiple son los plasmocitomas. Estos se presentan clínicamente como placas-nódulos eritematovioláceos o eritematoamarillentos, consecuencia de metástasis cutánea directa desde focos óseos adyacentes. Histológicamente se objetiva un infiltrado dérmico profundo e hipodérmico de células plasmáticas, que muestran positividad para CD38 y CD138 y negatividad para marcadores B. A diferencia de las lesiones por infiltración de MW, la presencia de plasmocitomas empeora el pronóstico de la enfermedad hematológica.
Destacamos la peculiaridad de nuestro caso debido a que la infiltración directa cutánea por el proceso linfoproliferativo es un hecho bastante infrecuente, según la literatura, y que no conlleva un empeoramiento en el pronóstico a pesar de ser una manifestación de progresión de la enfermedad.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


