


Un varón de 80 años consultó por lesiones cutáneas asintomáticas de 2 meses de evolución. No refería tratamientos habituales ni administración de fármacos nuevos, negaba fiebre u otra sintomatología acompañante y no recordaba haber sufrido ningún tipo de picadura.
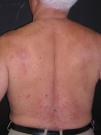
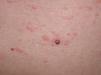
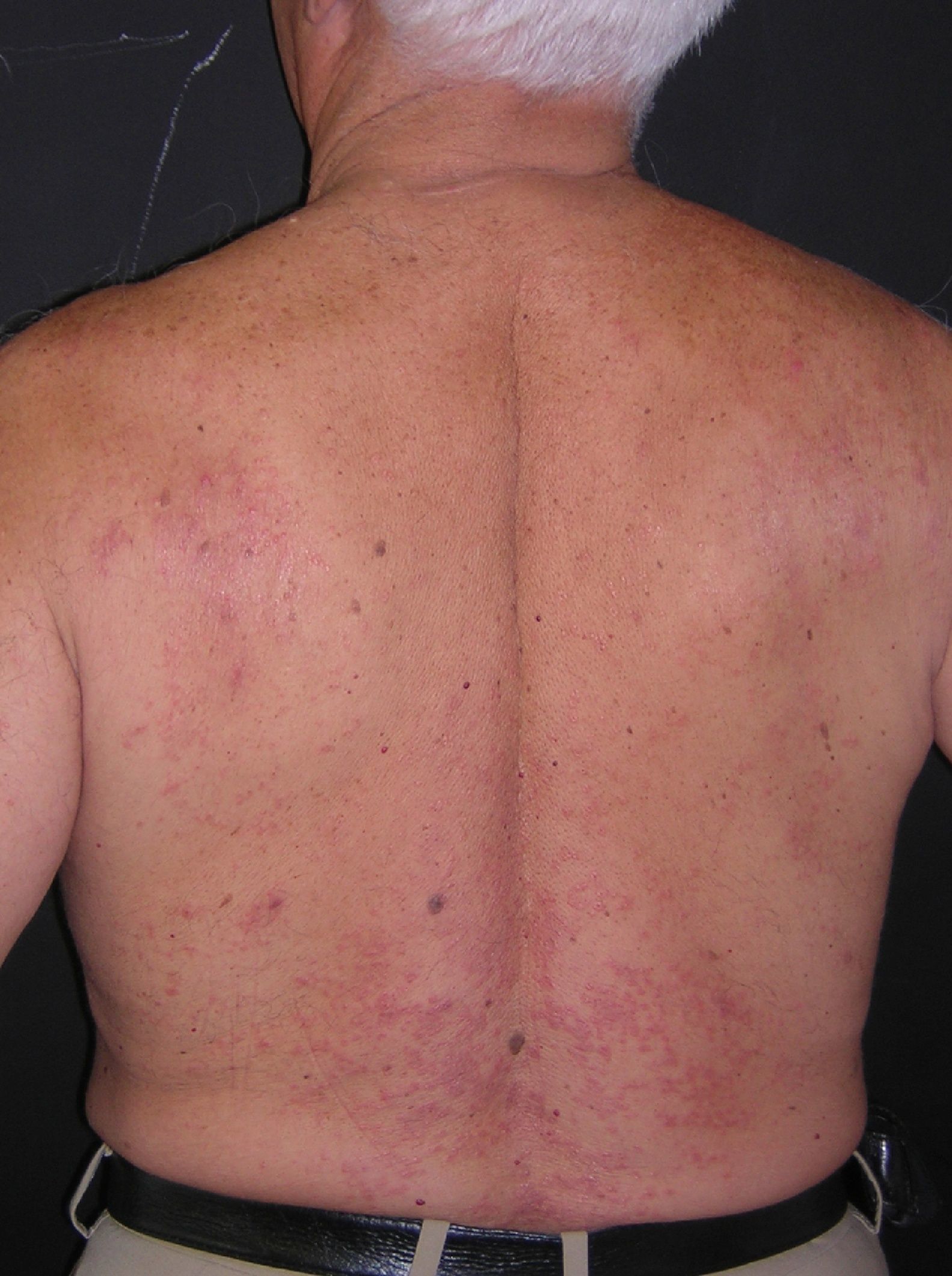
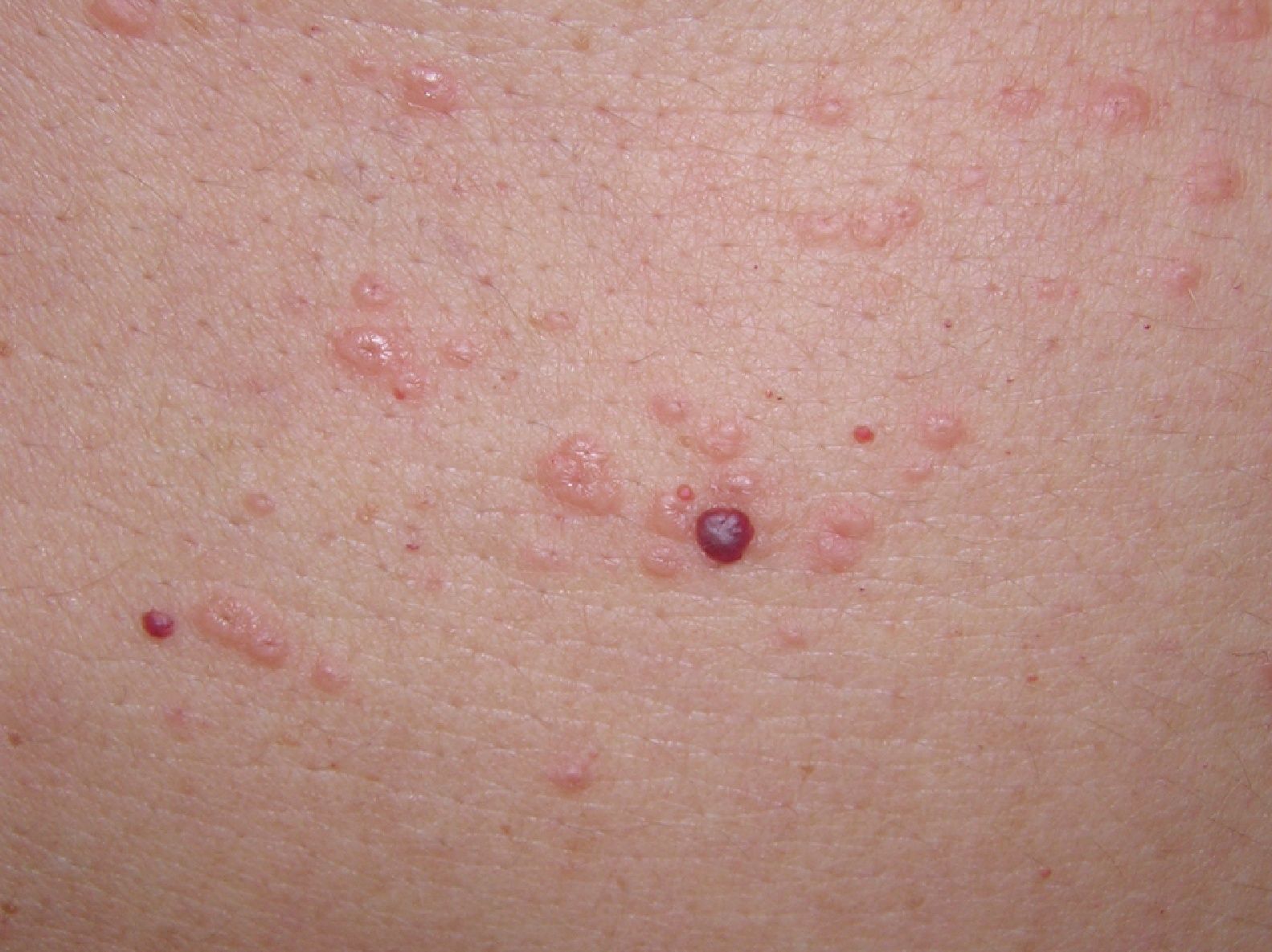
A la exploración se observaban en la parte superior del tronco pápulas eritematosas, de aproximadamente 3 mm de diámetro, de consistencia firme y centro deprimido (figs. 1 y 2). Algunas de ellas confluían formando placas. No había afectación de mucosas ni palmoplantar.
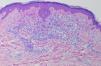
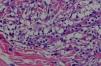
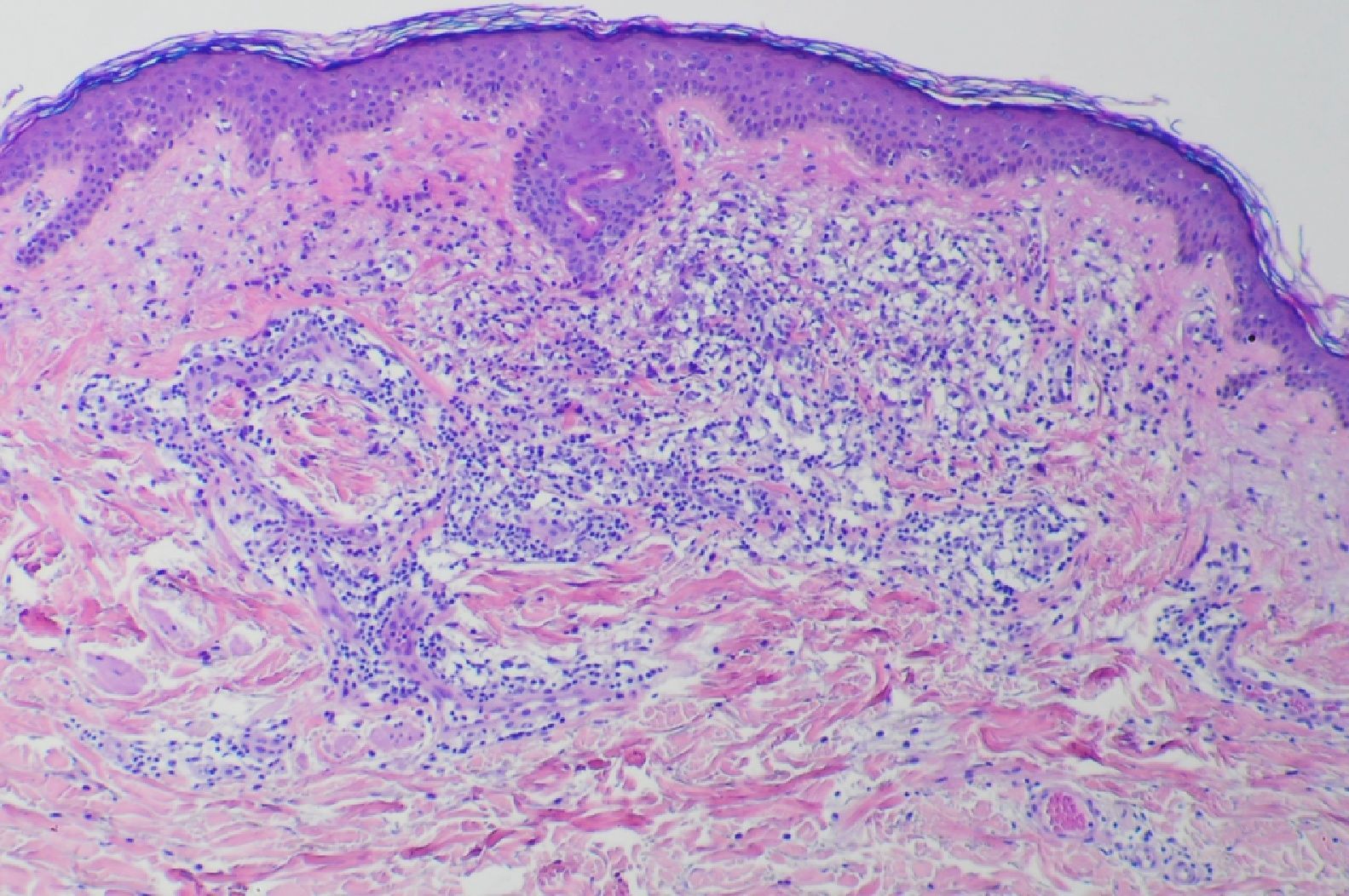
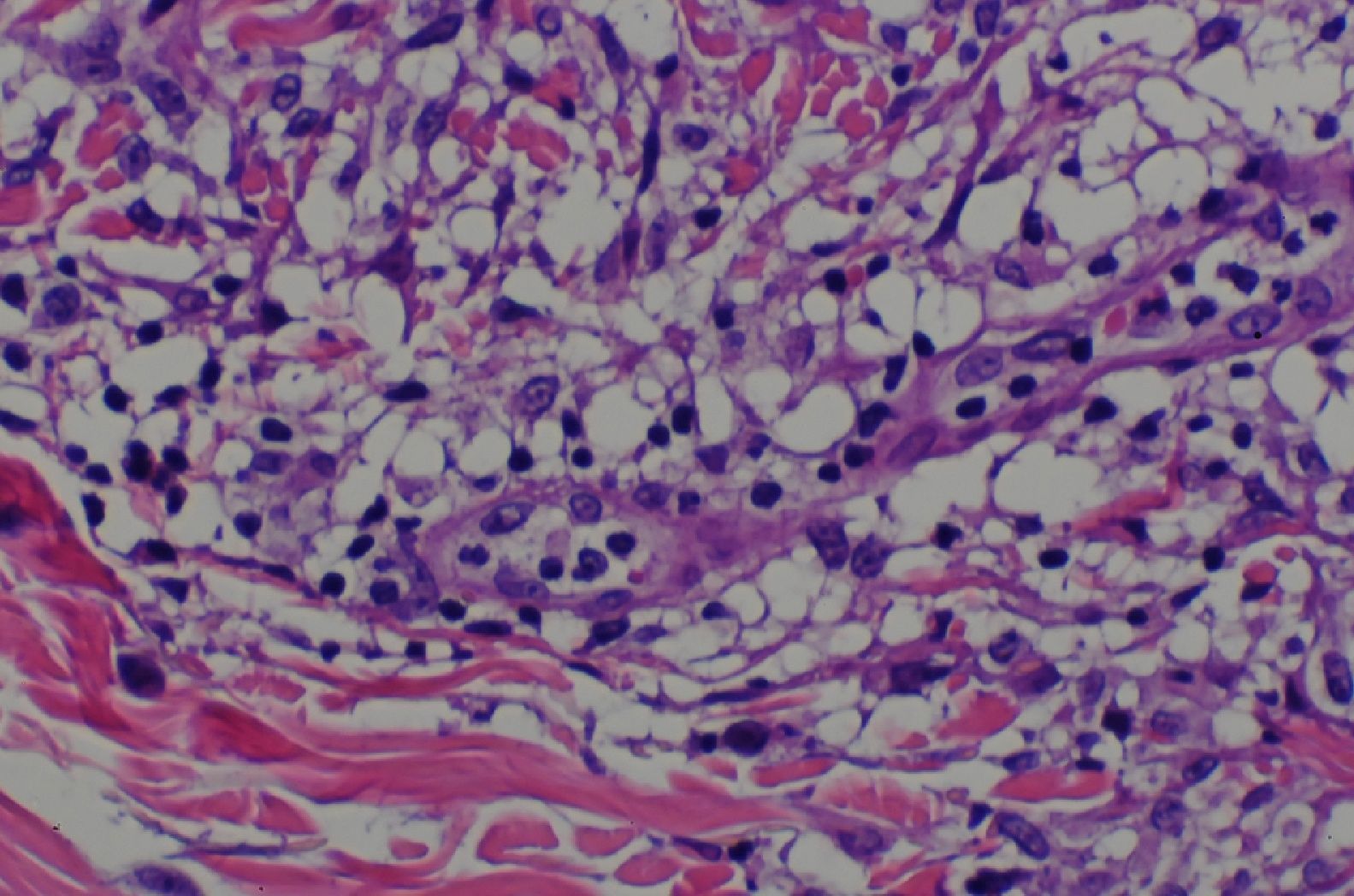
En la biopsia cutánea se observó un denso infiltrado ocupando la dermis papilar y media compuesto fundamentalmente por histiocitos, algunos de ellos vacuolados, linfocitos y alguna célula gigante multinucleada (figs. 3 y 4). El estudio inmunohistoquímico mostró positividad para CD68 y negatividad para S100, CD1a y FXIIIa.
En los estudios analíticos y radiológicos únicamente destacaba una monocitosis ya conocida y en seguimiento por el servicio de Hematología con el diagnóstico de leucemia mielomonocítica crónica (LMMC) con escasos rasgos displásicos en la médula ósea y cariotipo normal.
Se diagnosticó al paciente de histiocitosis eruptiva generalizada (HEG) y se adoptó una actitud expectante, desapareciendo las lesiones a los 6 meses del diagnóstico. La LMMC ha permanecido estable hasta la fecha.
Las histiocitosis son un grupo infrecuente y heterogéneo de enfermedades caracterizadas por la proliferación de células del sistema histiocitario. La mayor parte de los autores continúa utilizando la división clásica de: histiocitosis de células de Langerhans (HCL), histiocitosis tipo I o histiocitosis X e histiocitosis de células no Langerhans (HCNL), tipo II o no X.
Dentro del grupo de HCNL se incluyen múltiples enfermedades, como la HEG. Hay autores que defienden que no se trata de enfermedades independientes, sino que son estadios evolutivos de una misma enfermedad y que la HEG sería un estadio inicial del proceso, evolucionando posteriormente a lesiones de tipo xantomatoso1.
En 1963, Winkelmann y Muller describieron la HEG2 como un cuadro infrecuente caracterizado por múltiples pápulas eritematosas no confluyentes y simétricas distribuidas por el tronco y las extremidades. No se acompaña de afectación extracutánea e histológicamente destaca un infiltrado monomorfo compuesto predominantemente por histiocitos negativos para S100 y lípidos. Es un cuadro benigno que afecta fundamentalmente a pacientes de edad media. Tiende a la remisión espontánea y, aunque no se ha establecido la causalidad de esta, hay autores que defienden el mecanismo apoptótico cómo responsable3.
En nuestro paciente se planteó el diagnóstico diferencial clínico con el granuloma anular generalizado que fue descartado y mediante el estudio inmunohistoquímico se orientó el diagnóstico hacia un tipo de HCNL. Por el aspecto clínico de las lesiones, su localización y la edad de comienzo se descartan otras HCNL, como la histiocitiosis cefálica benigna, la reticulohistiocitosis multicéntrica o el histiocitoma nodular progresivo. El principal diagnóstico diferencial se plantea con otras HCNL de tipo xantomatoso, especialmente con el xantoma papular del adulto (XPA). Este XPA sería un estadio evolutivo posterior en el que las lesiones clínicas tendrían una coloración más amarillenta y que histológicamente se caracteriza por presentar más células xantomatosas4.
Generalmente, se mantiene una actitud expectante ya que es habitual la resolución espontánea en pocos meses. Cuando no existe remisión se plantea la necesidad de tratamiento, describiéndose buenos resultados mediante tratamiento sistémico con corticoides, hidroxicloroquina y talidomida5, con PUVA6 o incluso mediante el empleo de crioterapia, raspado o láser de dióxido de carbono en lesiones localizadas1.
Nuestro paciente presentaba 3 hechos singulares: la confluencia de las lesiones, la negatividad del FXIIIa y la asociación con una LMMC.
La confluencia de las lesiones se ha descrito tardíamente en esta entidad. Klemke et al.7 han relacionado esta característica clínica de esta histiocitosis con la leucemia monocítica aguda (LMA) y con un peor pronóstico de la enfermedad. Por eso, la presencia en nuestro paciente de lesiones confluentes y LMMC apoya esta observación.
Otro hecho característico de nuestro caso es la negatividad para el FXIIIa, que generalmente se había descrito positivo en las HEG, existiendo en la literatura otros 2 casos con esta característica6,8.
En general, las HCNL presentan un curso benigno en pacientes sanos y no suelen aparecer asociadas a enfermedades malignas. Sin embargo, algunas HCNL, como la histiocitosis nodular progresiva y el xantogranuloma juvenil, se han descrito asociadas a trastornos hematológicos9. Hasta el momento existen recogidos en la literatura 2 casos de HEG asociados a LMA7,10 pero ningún caso a LMMC, como sería el nuestro.
Como conclusión, presentamos el que creemos el primer caso de un paciente con HEG asociada a LMMC. En nuestra opinión, la frecuencia de aparición de trastornos hematológicos en pacientes con HCNL obliga a investigar esta asociación en pacientes con estos cuadros y más concretamente en HEG con lesiones confluyentes.


