




La histiocitosis de células de Langerhans (HCL) es un trastorno proliferativo clonal de células de Langerhans inmaduras1,2, que puede afectar a cualquier órgano3, ocurre predominantemente en niños, en adultos la incidencia es poco frecuente, siendo aún más raro la presentación limitada a la piel. Presentamos 3 casos de HCL cutánea en pacientes adultos, que se extirparon quirúrgicamente, sin evidenciar nuevas lesiones, afectación sistémica, ni otras alteraciones durante el seguimiento.
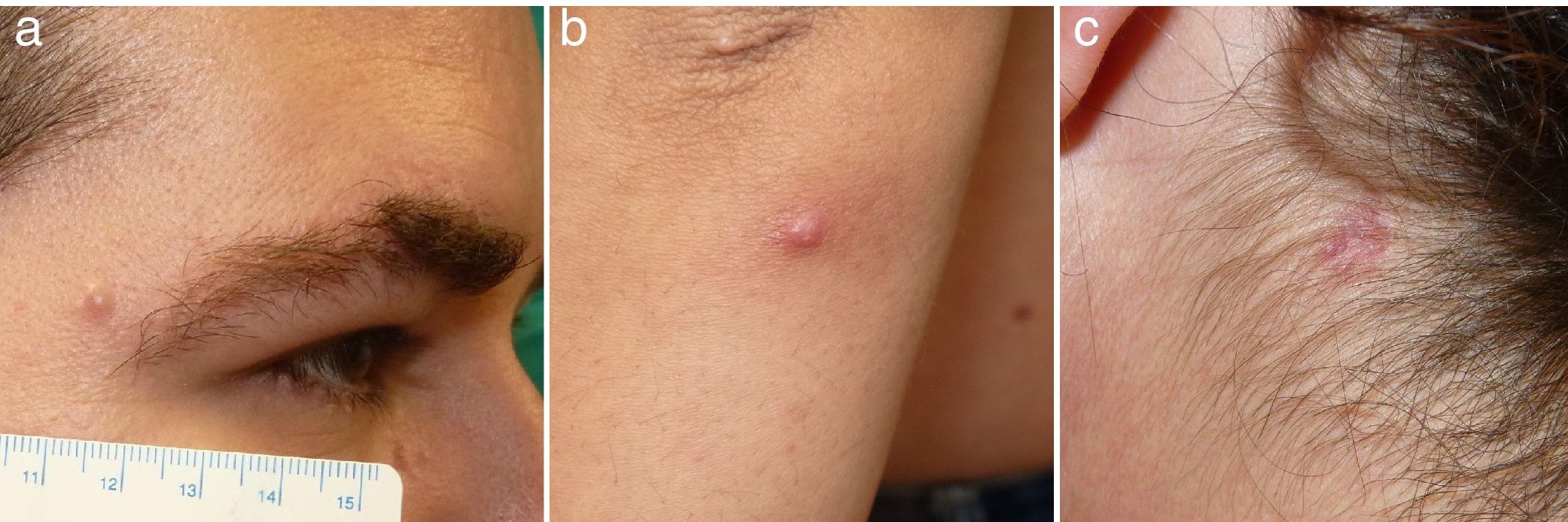
Caso 1: Varón de 36 años de edad, sin antecedentes de interés, que consultó por una lesión asintomática de 2 meses de evolución, localizada en borde externo de ceja derecha, sin otros síntomas asociados. A la exploración física se observó una pápula de 6mm de diámetro, levemente eritematosa, de consistencia firme y bien delimitada (fig. 1a); sin otras lesiones cutáneas ni mucosas; no se detectó adenopatías ni visceromegalias. El estudio histopatológico de la biopsia-extirpación reveló una epidermis sin alteraciones, y en la dermis un infiltrado celular denso con patrón nodular (fig. 2a) compuesto por células de núcleos ovalados-reniformes «en grano de café», con citoplasma claro eosinófilo, que correspondían a histiocitos. En el infiltrado asociaba zonas de necrosis y se acompañaba de linfocitos y eosinófilos (fig. 2b). El índice mitótico fue bajo. La inmunohistoquímica fue positiva para S100, CD1a (figs. 2c y d) y CD207 (langerina), y negativa para CD68. Con estos hallazgos se diagnosticó de HCL y se realizó estudió de extensión con hemograma, coagulación, bioquímica completa, perfil hormonal, estudio de orina, radiografía de tórax, ecografía abdominal y serie ósea radiológica, que resultaron normales. Finalmente se llegó al diagnóstico de HCL cutánea limitada del adulto. Tres años después continuó asintomático, no presentó nuevas lesiones cutáneas, ni afectación sistémica ni otras comorbilidades.
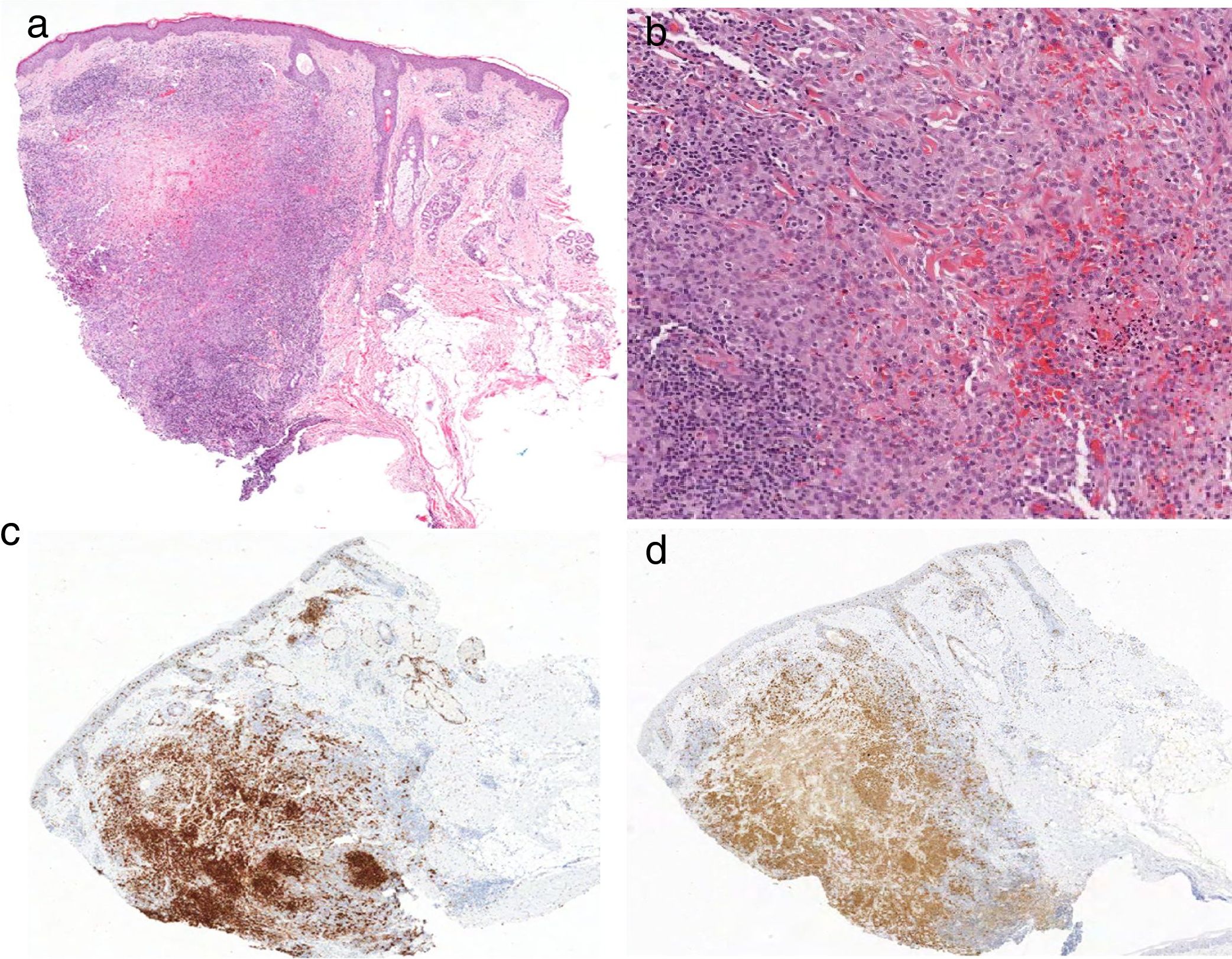
a) Infiltrado celular denso con patrón nodular en dermis; asocia en el infiltrado zonas de necrosis (hematoxilina-eosina, ×10); b) Histiocitos con núcleos ovalados-reniformes «en grano de café», con citoplasma claro eosinófilo, se acompaña de linfocitos y eosinófilos (hematoxilina-eosina, ×20). Inmunohistoquímica fue positiva para S100 (c) y CD1a (d).
Caso 2: Mujer de 33 años que consultó por una lesión asintomática en antebrazo izquierdo de 6 meses de evolución. Se trataba de una pápula eritematosa e infiltrada de 5mm (fig. 1b). No presentaba adenopatías ni visceromegalias. Se extirpó la lesión, y los hallazgos histopatológicos fueron similares a los descritos del caso 1, compatible con HCL. El estudio de extensión fue negativo, y a los 2 años de seguimiento no desarrolló nuevas lesiones ni otras alteraciones clínicas ni analíticas.
Caso 3: Mujer de 49 años, consultó por 2 lesiones papulares eritematosas de 5 y 7mm, localizadas en mejilla derecha, espalda, y una placa de 9mm en cuello lateral izquierdo (fig. 1c). No se detectaron alteraciones en la exploración física. El estudio histopatológico de una lesión fue compatible con HCL, por lo que se realizó extirpación de las 3 lesiones, mostrando los mismos hallazgos. El estudio de extensión también fue negativo. No se evidenció nuevas lesiones ni otra comorbilidad durante los 3 años posteriores de seguimiento.
La HCL es una enfermedad poco frecuente, ocurre mayoritariamente en menores de 15 años, con una incidencia estimada de 5-9 casos por millón, y de uno en un millón para mayores de esta edad1,4. La HCL incluye un amplio espectro de manifestaciones clínicas3–6, y varían de acuerdo al órgano afectado, desde lesiones solitarias autorresolutivas hasta enfermedad diseminada con compromiso vital3,4,6,10. Actualmente se encuentra clasificada dentro del grupo de histiocitos del grupo L, propuesto por la sociedad de histiocitosis4. El diagnóstico se basa en hallazgos clínicos y radiológicos, en combinación con los histopatológicos, que identifican la infiltración de histiocitos con un inmunofenotipo característico. La enfermedad puede afectar cualquier órgano, fundamentalmente los huesos, la piel, la hipófisis, el hígado, el bazo, los ganglios linfáticos, los pulmones; y con menos frecuencia, el sistema nervioso central (excepto la hipófisis) y hematopoyético3,4.
La patogénesis permanece aún sin aclarar3,4,6,7; la mutación del gen BRAF V600E, que da lugar a una sobre estimulación de la vía MAPK se encuentran presente en el 50% de los pacientes con HCL; en un menor porcentaje se encuentran mutaciones en el MAP2K1 (MEK1), otras mutaciones de esta vía incluyen la MAP3K1, ARF. También se han descrito mutaciones en la vía del PIK3CA, PICK1, PICK3R24. Estos hallazgos se encuentran a favor que la HCL es un proceso neoplásico3,4, sin embargo el componente inflamatorio y el curso ocasionalmente benigno, incluso con regresión espontánea, sugieren un proceso reactivo. Se ha publicado además, casos familiares de HCL sin identificar hasta ahora una susceptibilidad genética4.
La HCL limitada a la piel en el adulto es una presentación muy rara1,5,8. Puede presentarse como una lesión solitaria (papular o nodular, con o sin ulceración) o múltiples1,9; incluso simulando otras dermatosis10. En una serie de 18 pacientes con HCL, y de presentación cutánea inicial, con edades comprendidas entre 20-89 años, sin predilección por la raza o el género, cerca del 50% (8/18) fueron lesiones únicas, tipo pápulas y nódulos, y como lesiones ulceradas en pliegues1. El estudio de extensión, uno presentó afectación ósea y 2 asociaban un síndrome mielodisplásico. Durante el seguimiento (media de 41 meses), 2 pacientes desarrollaron lesiones extracutáneas de HCL y 5 neoplasias hematológicas con mala evolución: leucemia mielomonocítica aguda en 2 casos, sarcoma histiocítico, linfoma de células B grande difuso y linfoma de células T periférico. Además, los autores revisan la literatura de 74 pacientes con presentación inicial en la piel, 4 presentaron extensión de la enfermedad y 8 alteraciones hematológicas durante el diagnóstico o seguimiento1. La asociación de la HCL y los trastornos mielomonocíticos puede estar relacionado a un origen común de las células en la médula ósea1. En la actualidad se considera que la HCL se origina de células dendríticas mieloides de la médula ósea, que expresan los mismos antígenos que las células de Langerhans cutáneas.
Dentro del diagnóstico diferencial se incluye diversos procesos neoplásicos e inflamatorios10, por lo que el estudio histopatológico con inmunofenotipo es fundamental para realizar el diagnóstico, demostrando la infiltración de células de Langerhans con la tinción CD1a+ y/o CD207+ (langerina)3,6, lo cual ha reemplazado a la identificación de los gránulos de Birbeck mediante microscopia electrónica6. La tinción CD207+ permite realizar el diagnóstico diferencial con la histiocitosis de células indeterminadas, donde es negativa4.
Es importante el estudio de extensión y el seguimiento de los pacientes adultos con lesiones cutáneas para descartar afectación de HCL en otras localizaciones y descartar comorbilidades1,6, principalmente hematológicas, como suele ocurrir en los niños. En el manejo de la HCL en adultos se recomiendan estudios de laboratorio y de imagen basales (hemograma, bioquímica, radiografía de tórax, hormonas, serie ósea y ecografía abdominal) y durante el seguimiento1,3, que se suele recomendar cada 6 meses en los pacientes que permanecen asintomáticos3, con exploración física completa, hemograma, bioquímica y ecografía abdominal; además de radiografía de tórax anuales, durante al menos los 3 primeros años3. La biopsia de médula ósea debería realizarse si se sospecha infiltración, aunque algunos autores la recomiendan de rutina1.
El tratamiento óptimo de la HCL en el adulto, al ser tan infrecuente, no está establecido. Los pacientes con HCL limitada, con compromiso de un solo órgano o sistema, presenta buen pronóstico3,6, pero es recomendable un seguimiento estrecho en todos los casos3,10. Se puede realizar tratamiento local (cirugía, corticoides tópicos e infiltración de corticoides); en el caso de lesiones múltiples, ulceradas o resistentes se suele emplear corticoides sistémicos, fototerapia, radioterapia, interferón y diversos esquemas de quimioterapia6,9. Se sugirió que la ulceración de las lesiones puede influir en el pronóstico, aunque hasta ahora existen resultados contradictorios1.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Al Dr. Juan Luis Santiago Sánchez-Mateos, y a la Dra. Claudia Ramos-Rodríguez, por su colaboración en este trabajo.


