



La histiocitosis de células indeterminadas (HCI) es un cuadro infrecuente caracterizado por la proliferación de histiocitos de fenotipo inmunohistoquímico mixto entre células de Langerhans e histiocitos no Langerhans. Su comportamiento varía entre formas sistémicas agudas fulminantes y formas solo cutáneas de curso benigno crónico o autorresolutivo.
Presentamos el caso de una niña de 16 meses de edad, sin antecedentes personales ni familiares de interés, que acude a consulta por la aparición brusca de lesiones faciales de 15 días de evolución, sin mejoría con amoxicilina-clavulánico pautado por su pediatra de zona ante el inicio brusco del cuadro. La anamnesis no reveló signos ni síntomas de enfermedad asociada. La exploración física mostraba múltiples pápulas de predominio facial eritemato-anaranjadas con un halo eritematoso, algunas con tendencia a la seudovesiculación, umbilicación y formación de costra central (fig. 1A), sin adenopatías palpables. Se procedió a tomar un frotis de las lesiones para visión directa, cultivo de microorganismos y PCR del virus varicela-zóster y Coxsackie. Los resultaron fueron normales, y una semana después todas las lesiones persistían en fase papular, asociando la aparición de pápulas amarillo-anaranjadas en antebrazos y raíz de miembros (fig. 1B y C). La paciente mantenía un buen estado general. Con el diagnóstico de sospecha de histiocitosis cefálica benigna, eruptiva generalizada o histiocitosis de células de Langerhans (HCL), se procedió a realizar una biopsia cutánea de una de las lesiones del antebrazo derecho. La biopsia mostraba una epidermis ulcerada con un infiltrado de predominio mononuclear-histiocitoide, con linfocitos y algunos eosinófilos acompañantes, tanto en dermis papilar como en dermis profunda, donde formaban nódulos histiocitarios (fig. 2). La inmunohistoquímica fue difusa e intensamente positiva para CD1a tanto en la epidermis como en los nódulos dérmicos, con clara positividad intensa para CD68 y S100, aunque de forma más dispersa tanto en la epidermis, dermis intersticial y en los nódulos histiocitarios. Por el contrario, la langerina (CD207) aparecía positiva en el control epidérmico, y de forma muy focal, prácticamente negativa, en dermis intersticial y nódulos dérmicos. Con estos hallazgos realizamos el diagnóstico de HCI (fig. 3). Se completó el estudio con bioquímica, hemograma, osmolaridad en plasma y sedimento de orina, junto a una ecografía de abdomen, serie radiográfica de tórax, calota y columna vertebral. Las exploraciones complementarias estaban dentro de la normalidad. Tras 2 meses de evolución las lesiones faciales comenzaron a resolverse estando la paciente únicamente en tratamiento con metilprednisolona tópica. Sin embargo aparecieron lesiones ampollosas en ambos antebrazos y manos (fig. 1D), que obligaron a iniciar tratamiento sistémico con corticoides con buen control de las mismas. Hasta la fecha no han aparecido complicaciones sistémicas, pero la paciente requiere dosis bajas de mantenimiento de corticoides orales para evitar brotes de lesiones vesículo-ampollosas en tronco y extremidades, junto a controles periódicos por la posibilidad de desarrollar neoplasias hematológicas asociadas o formas extracutáneas y multiorgánicas de su enfermedad.
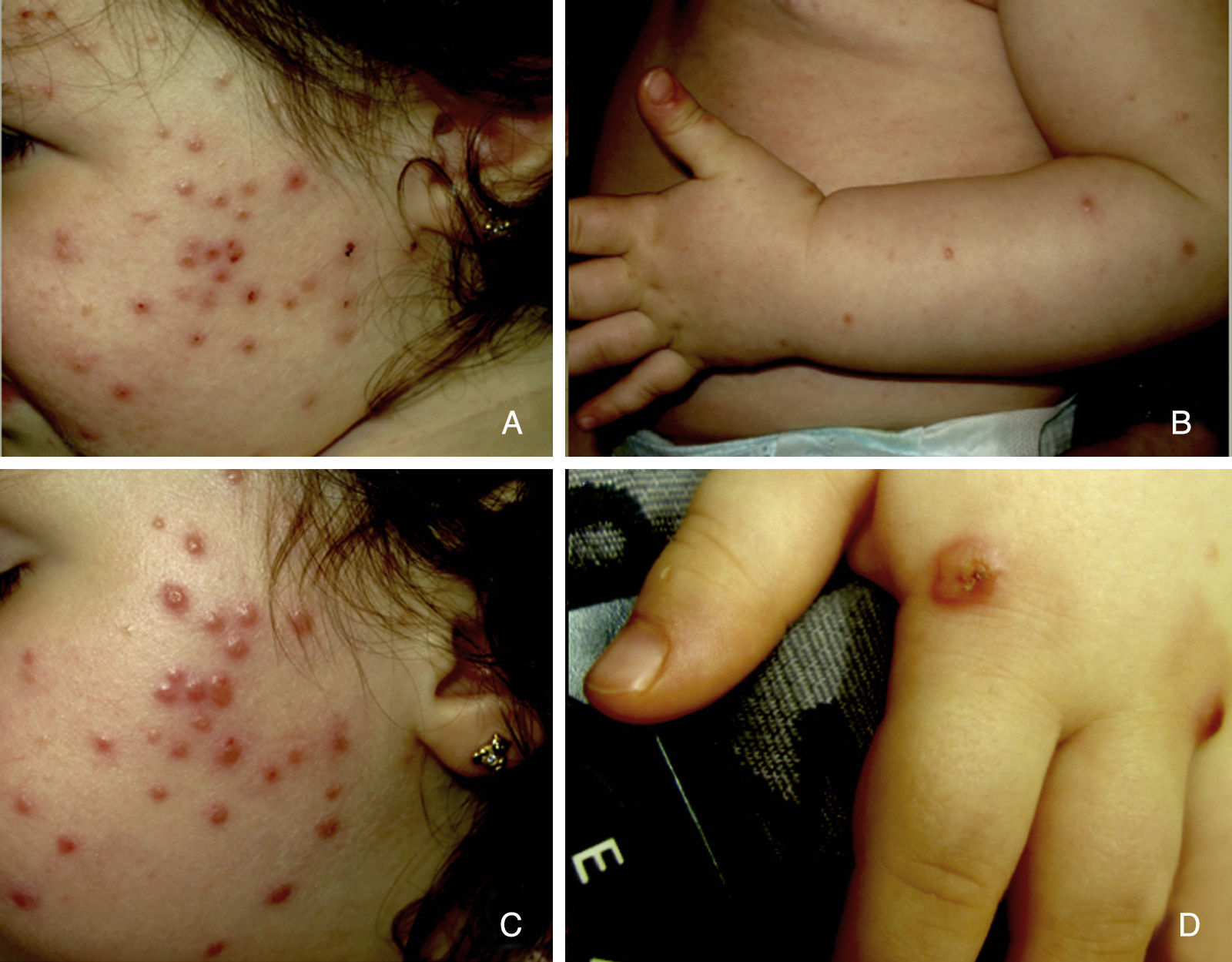
A) Pápulas eritemato-anaranjadas, algunas con umbilicación, seudovesiculación y formación de costra. B y C) Una semana después, todas las lesiones en fase papular con aparición de pápulas amarillo-anaranjadas en antebrazos. D) Lesiones ampollosas en manos, 2 meses después del comienzo del cuadro.
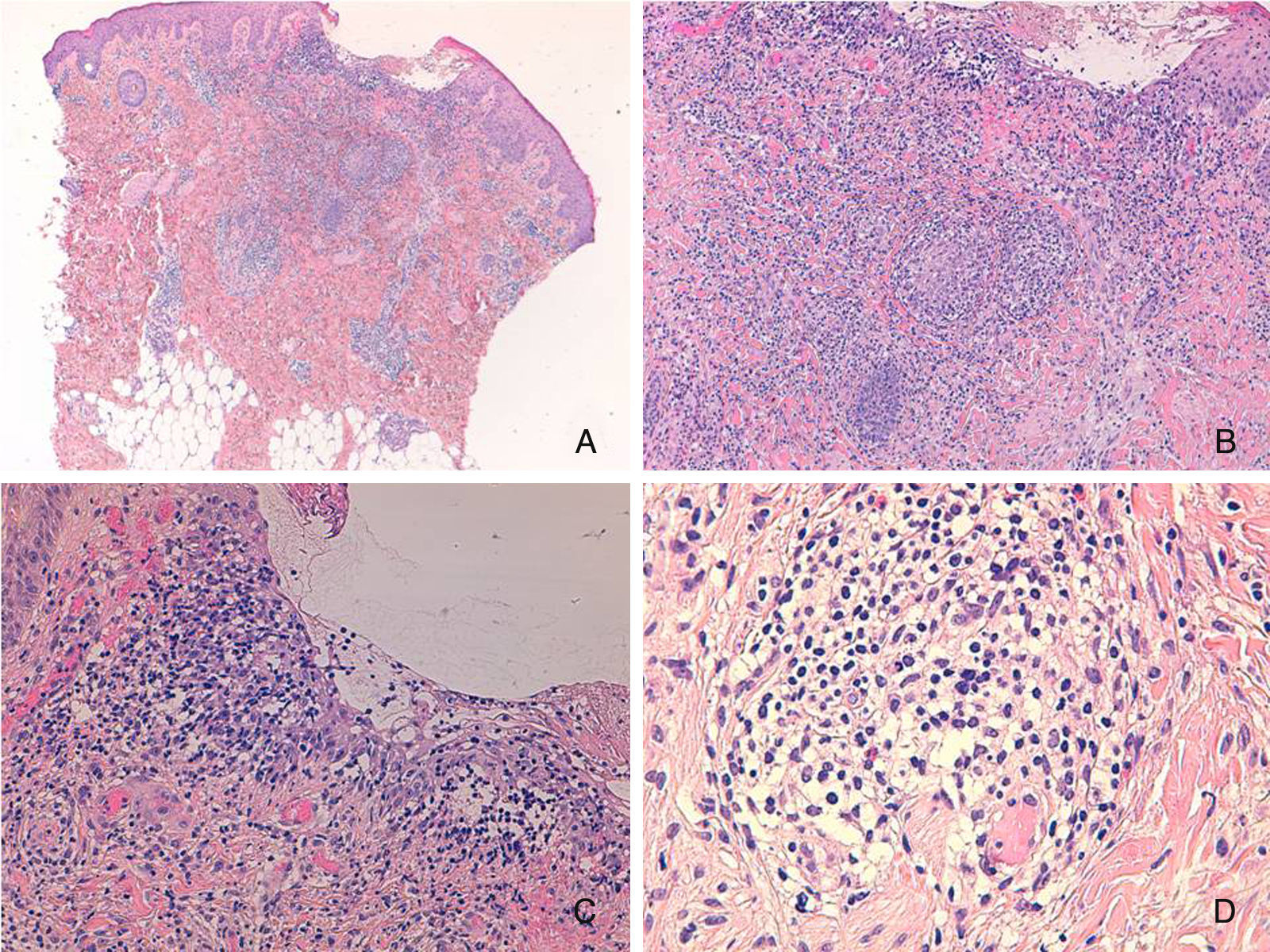
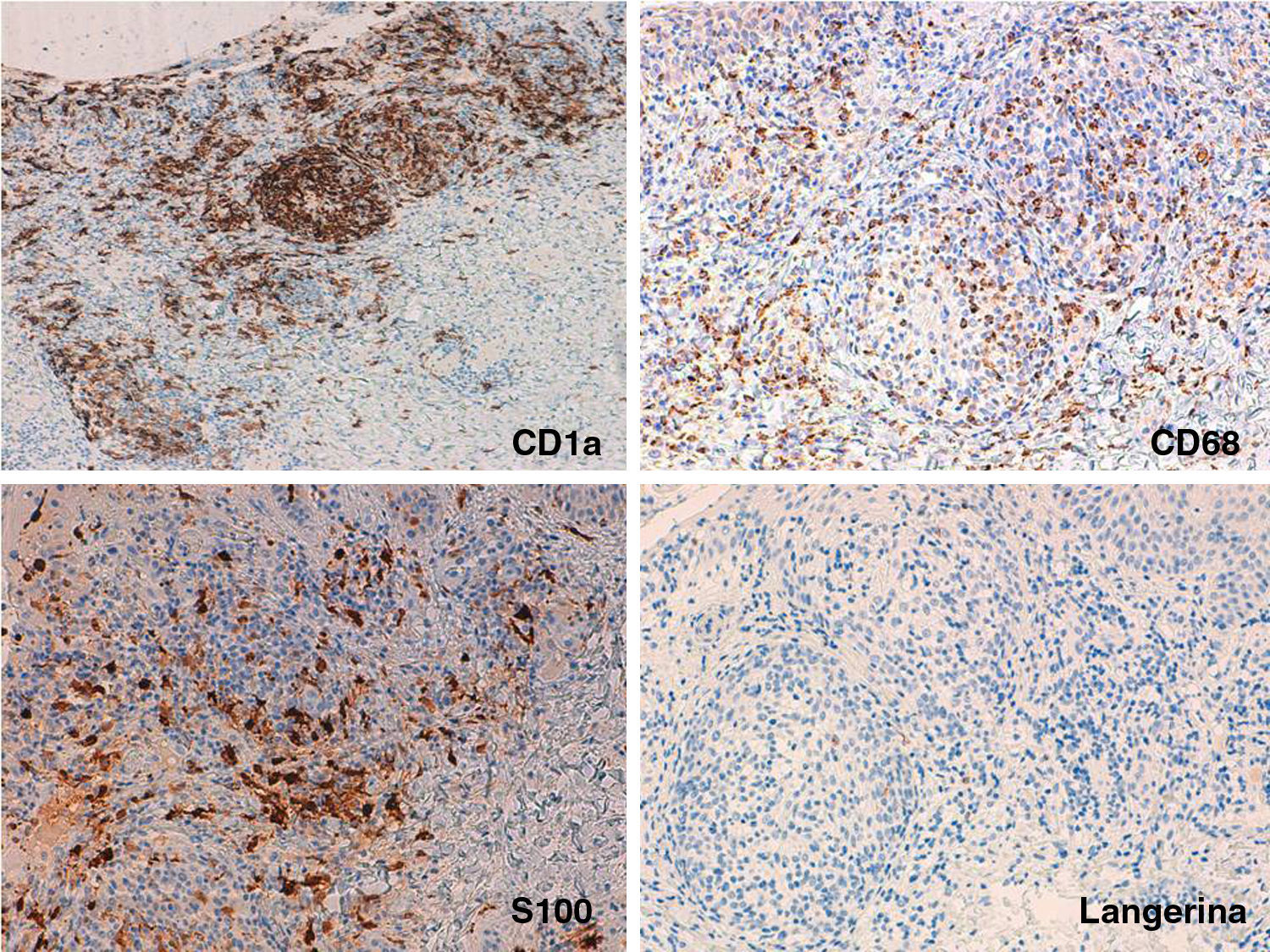
Inmunohistoquímica: A) ×100 con intensa y difusa positividad para CD1a. B y C) Detalle de los nódulos a ×200 con positividad dispersa pero intensa para CD68 y S100 en epidermis, dermis intersticial y nódulos dérmicos. D) Detalle de los nódulos a ×200 con escasa positividad para langerina, en las mismas zonas en las que CD1a, CD68 y S100 eran positivos.
Las histiocitosis son un grupo heterogéneo de trastornos que tienen en común una proliferación excesiva de histiocitos. Clásicamente se dividen según su fenotipo inmunohistoquímico en HCL: CD1a+, S100+, langerina+ con CD68−, y en histiocitosis no-Langerhans: CD68+, CD1a−, S100−, langerina− y factor XIIIa±. La HCI comprende un grupo poco frecuente dentro de este espectro con marcadores de los 2 grandes grupos, esto es CD1a+, S100+, CD68+, siendo la langerina−, marcador inmunohistoquímico de los gránulos de Birbeck1,2.
La HCI se ha descrito en cualquier rango de edad, incluso de forma congénita. Se presenta con lesiones papulares rojo-marronáceas en tronco y extremidades que pueden ulcerarse, con distribución generalizada o como una lesión aislada, existiendo casos reportados similares al nuestro con predominio o exclusividad de lesiones faciales1. Aunque tiene tendencia por la afectación exclusiva cutánea en forma de cuadros agudos, crónicos o remitente-recurrentes de curso indolente e incluso formas autorresolutivas, ocasionalmente puede existir afectación ocular, ósea e incluso multiorgánica con curso letal2. Por ello su patrón de presentación clínica puede simular tanto una HCL como no-Langerhans, sobre todo formas eruptivas generalizadas e histiocitosis cefálica benigna, siendo el estudio histológico imprescindible para identificar la entidad3.
En cuanto a la fisiopatogenia, Badalian-Very et al. en 2010 describieron una serie de 61 pacientes con HCL con un 57% de mutaciones en BRAF V600E4, y recientemente se han descrito 3 casos de HCI con translocaciones en los genes ETV3-NCOA2 no encontradas en otros casos de histiocitosis, siendo NCOA2 el codificador de la proteína TIF2, un cofactor transcripcional de receptores nucleares y esteroideos5. Estos datos apoyarían el origen clonal de las histiocitosis, además de que en un futuro puedan tener implicaciones terapéuticas. Sin embargo existen en la literatura cuadros de histiocitosis con perfil inmunohistoquímico de HCI, aparecidos tras procesos inflamatorios como la escabiosis o pitiriasis rosada con curso autorresolutivo6,7. Esto nos permite plantear la hipótesis de que nos encontramos frente a espectros de enfermedades solo definidas por su patrón inmunohistoquímico, que incluirían cuadros realmente neoplásicos y cuadros inflamatorios reactivos. Probablemente, el estudio molecular pueda contribuir en un futuro cercano a una mejor caracterización de esta entidad.
En cuanto al tratamiento de la HCI, dada la baja frecuencia de esta enfermedad, no existen fármacos claramente de elección. La conducta expectante es una opción dado el curso indolente o incluso autorresolutivo en la mayoría de los casos, ayudándose en las formas cutáneas de corticoides tópicos. No obstante, en la literatura se han reportado una gran variedad de tratamientos diferentes a los corticoides sistémicos como la fototerapia, talidomida, isotretinoína, metotrexato, ciclofosfamida, pravastatina, baño de electrones o exéresis de lesiones solitarias8,9. La poliquimioterapia es una opción que generalmente se reserva para los casos más severos multiorgánicos, pudiendo considerarse, igual que en el resto de neoplasias hematológicas, el trasplante de progenitores hematopoyéticos en cuadros refractarios y que supongan una amenaza para la vida del paciente.
Para concluir, queremos incidir en la relevancia del control evolutivo de estos pacientes, para detectar precozmente la posibilidad de afectación visceral por la propia enfermedad o la aparición de segundas neoplasias hematológicas asociadas. En este sentido, los linfomas B de bajo grado y las leucemias agudas han sido los más reportados en la literatura, incluso años después del comienzo de la HCI, lo que nos obliga a un control clínico y analítico prolongado10.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


