







El hamartoma fibroso de la infancia (HFI) es tumor poco frecuente de tejidos blandos en lactantes y niños, caracterizado histopatólogicamente por su morfología trifásica y curso benigno.
MétodosPresentamos las características histopatológicas e inmunohistoquímicas de una serie de 21 casos de HFI y revisamos sus principales diagnósticos diferenciales.
ResultadosLas lesiones predominaron en varones con una edad media de 3,7 años (5 meses-24 años) en localizaciones previamente descritas. Histopatológicamente, se caracterizaron por proporciones variables de tejido fibroblástico, mesenquimal y adiposo maduro. Tres casos (21%) mostraron zonas hialinizadas con artefactos en grietas, que imitaban al fibroblastoma de células gigantes. Presentaron tinción positiva para actina de músculo liso 8/13 (61%) en el componente fibroblástico, CD34 en 6/8 (75%) en el mesenquimal inmaduro y fibroblástico; y S100 en el tejido adiposo en 7/7 (100%).
ConclusiónNuestros hallazgos histopatológicos son superponibles a los descritos en series amplias. No obstante, la heterogeneidad clínica obliga a conocer las variantes de esta entidad dentro de su típica histomorfología trifásica, para lograr un correcto diagnóstico y manejo terapéutico.
Fibrous hamartoma of infancy (FHI) is a rare soft-tissue tumor usually seen in infants and young children. Histologically, the tumor is characterized by a triphasic morphology, and the clinical course is benign.
MethodsWe described the histopathologic and immunohistochemical features in a series of 21 cases of FHI and reviewed the main entities to consider in differential diagnosis.
ResultsMost patients in the series were male. The mean age was 3.7 years (range, 5 months–24 years), and lesions were found in locations that have been previously reported. Histopathologically, the tumors were composed of variable proportions of fibroblastic, mesenchymal, and mature adipose tissue. Three of the 13 immunohistochemically stained biopsies (14%) contained hyalinized zones with cracking artifacts, mimicking giant cell fibroblastoma. Eight of the 13 stained with smooth-muscle actin (61%) were positive in the fibroblastic component, 6 of the 8 stained with CD34 (75%) were positive in the immature mesenchymal and fibroblastic components, and all 7 stained with S100 were positive (100%) in adipose tissue.
ConclusionsOur histopathologic findings are consistent with those described in larger series. However, in order to reach a precise diagnosis and plan treatment, clinical heterogeneity obliges us to become familiar with variations in the characteristic triphasic histology of FHI.
La Organización Mundial de la Salud (OMS) define el hamartoma fibroso de la infancia (HFI) como un tumor pediátrico de tejidos blandos, benigno, pobremente delimitado, caracterizado por una histología trifásica compuesta por fascículos fibroblásticos, tejido adiposo maduro y nódulos de células mesenquimales inmaduras en un estroma mixoide primitivo1. Fue descrito por primera vez en 1956 por Reye et al. como un «fenómeno reactivo», que denominó tumor subdérmico fibromatoso de la infancia2 y posteriormente, renombrado como HFI por Enzinger et al. basados en su presentación clínica y sus características hamartomatosas3. A partir del 2005, comienzan a notificarse casos aislados con alteraciones citogenéticas complejas: t (6; 12; 8) (q25; q24.3; q13), t (2; 3) (q31; q21), y reordenamientos cromosómicos: (1, 2, 4 y 17)4–6. Sin embargo, no es hasta el 2016 cuando Park et al. describen, en 12 de sus casos, una mutación tipo inserción/duplicación en el exón 20 del receptor del factor de crecimiento epidérmico (EGFR), sugiriendo que se trata de un proceso neoplásico7. Posteriormente, Ellington et al. comentan un nuevo caso con dicha mutación8. Al-Ibraheemi et al. realizaron estudios genómicos por microarray en ocho de sus casos, y los dos que mostraron características histológicas sarcomatosas revelaron cambios en el número de copias genómicas. Ambos presentaron cariotipos moleculares complejos (hiperdiploide/tetraploide): uno, con una pérdida de heterocigosidad de las regiones cromosómicas 1 p y 11p, y el otro con pérdida del cromosoma 14 y de los fragmentos 10p, 22q119. La identificación de las mismas alteraciones citogenéticas en los distintos componentes de la lesión apoyan la consideración del HFI como un proceso neoplásico, clasificación con la que estamos de acuerdo.
Pese a que no existen asociaciones reconocidas del HFI con ningún trastorno familiar o sindrómico, hemos encontrado un caso relacionado con esclerosis tuberosa10 y otro con síndrome de Williams (SW)11. En el segundo, al encontrarse una mínima expresión de elastina dentro del tumor, los autores hipotetizan que una microdeleción en el gen de la elastina puede predisponer al desarrollo de HFI11.
El HFI es una entidad clínicamente heterogénea que afecta a localizaciones muy variadas. Habitualmente, se presenta en forma de nódulo único mal delimitado de unos 0,5 a 10 cm de diámetro sin afectación epidérmica12. No obstante, se han descrito variantes clínicas como multicéntrico13,14, gigantes (de hasta 20 cm)15, con hiperpigmentación16, hipertricosis e hiperhidrosis17–19.
Generalmente, son asintomáticos, pero pueden presentarse como nódulos infiltrados o adheridos a planos profundos, dificultando así el diagnóstico diferencial con una neoplasia maligna20. Sus localizaciones más habituales son la axila, la zona alta de la espalda, los brazos y el área genital9,21,22. Presentan un crecimiento acelerado en los cinco primeros años de vida con posterior estabilización. No se han descrito pacientes con involución o metástasis, pero sí dos casos con áreas sarcomatosas. El tratamiento de elección es la escisión quirúrgica completa con bordes libres22. Las cifras de recidiva publicadas se sitúan en torno al 15%15,16,23-26, si bien, en centros especializados estas pueden disminuir hasta el 1%9.
La inmunohistoquímica del hamartoma fibroso de la infancia es inespecífica y similar a otros tumores. El componente fibroblástico es típicamente positivo para vimentina y actina de músculo liso, pero también puede revelar reactividad focal para cúmulo de diferenciación 68 (CD68), desmina y factor XIIIa, pero no para S100, marcador que sí teñirá el tejido adiposo. En cuanto al componente inmaduro, muestra característicamente positividad para cúmulo de diferenciación 34 (CD34)21,25.
Describimos las principales características histopatológicas e inmunohistoquímicas (IHQ) de 21 casos de HFI y revisamos sus principales diagnósticos diferenciales en función de su patrón predominante.
Materiales y métodosSe diseñó un estudio observacional retrospectivo que incluyó los casos diagnosticados de HFI de entre los años 1990 y 2015 en el Centro Médico Voth, el Departamento de Dermatopatología del Hospital Universitario de la Princesa de Madrid y el Friedrichshafen Dermatopathologie, en Friedrichshafen (Alemania). En cada caso, el tejido se fijó en formalina tamponada al 5%, se procesó habitualmente y se incluyó en parafina. Se tiñeron secciones de 4 μm con hematoxilina y eosina y, para los estudios con IHQ, se montaron secciones de 4 μm en portaobjetos con carga positiva. Estas se dejaron secar más tarde durante la noche a 45°C. Los portaobjetos se desparafinaron en xilol durante 30 minutos, se rehidrataron usando concentraciones graduadas de etanol y se incubaron durante 30 minutos a 95°C en tampón ácido etilendiaminotetraacético (EDTA) (pH = 9,0). Se enfriaron a temperatura ambiente durante 20 minutos. Después del enfriamiento rápido con bloqueo de fosfatasa alcalina y biotina, usando avidina, las secciones se incubaron con los diferentes anticuerpos utilizados (tabla 2).
Resultados de tinciones inmunohistoquímicas
| Caso | S100 | Actina | PGM1 | CD34 | CD31 | Podoplanina |
|---|---|---|---|---|---|---|
| 1 | A | F | ± | |||
| 2 | A | - | M++ | + Vasos | ||
| 3 | F | F+ | ||||
| 4 | - | ± | M +A+ | + Vasos | + Vasos | |
| 5 | F | ± | - | |||
| 6 | - | ± | F+M+ | +Vasos | + Vasos | |
| 7 | A | |||||
| 8 | F | ± | - | |||
| 9 | A | F | ± | |||
| 10 | A | F | ||||
| 11 | ||||||
| 12 | A | F | F+ | |||
| 13 | A | F | M+ | + Vasos | +Vasos | |
| 14-21 | NA | |||||
| Total n (%) | 7 (100%) | 8 (61%) | 6 (100%) | 6 (75%) | 4 | 3 |
CD31: cúmulo de diferenciación 31; CD34: cúmulo de diferenciación 34; NA: No aplica; PGM1: fosfoglucomutasa-1.
La positividad para un determinado tejido se marca con la letra correspondiente (A: adiposo; F: fibroblástico; M: mesenquimal).
Las tinciones que no muestran predominancia por un determinado tejido se marcan con los signos: +: positivo; –: negativo; ±: leve.
Aquellas muestras que no fueron teñidas para un determinado anticuerpo no se han completado.
Los casos testados para desmina (2), NKIC3 (2), EMA (3) y la expresión nuclear de beta-catenina nuclear (1) no se han incluido.
Esta revisión ha sido aprobada por un comité de ética en investigación y se llevó a cabo de acuerdo con los principios éticos de la Declaración de Helsinki.
ResultadosCaracterísticas clínicas y epidemiológicasLas biopsias pertenecían a 13 hombres y ocho mujeres. La mediana de edad al diagnóstico de las lesiones era de un año (rango de cinco meses a 24 años). Estas se presentaban localizadas en sitios típicos (axila, miembro superior, región dorsal y cervical posterior) en 18 de nuestros pacientes y en sitios atípicos (cadera, mano, perianal) en los otros tres (tabla 1).
Características clínicas e histológicas de la serie de pacientes con HFI. Predominancia de los componentes según si representa igual o más del 45% de toda la lesión: n (%)
| Características clínicas | Características histológicas | |||||
|---|---|---|---|---|---|---|
| Caso | Sexo | Edad (años) | Localización | Tejido fibroblástico % | Tejido adiposo maduro % | Tejido mesenquimal % |
| 1 | Varón | 2 | Axila | 30 | 65 | 5 |
| 2 | Mujer | 24 | MS | 5 | 55 | 40 |
| 3 | Varón | 2 | MS | 25 | 70 | 5 |
| 4 | Varón | 0,5 | Hombro | 10 | 50 | 40 |
| 5 | Varón | 1 | Cadera | 5 | 70 | 5 |
| 6 | Mujer | 13 | MS | 8 | 90 | 2 |
| 7 | Mujer | 1 | MS | 45 | 35 | 20 |
| 8 | Varón | 1 | MS | 40 | 50 | 10 |
| 9 | Varón | 1 | Espalda | 35 | 35 | 30 |
| 10 | Mujer | 8 | Espalda | 30 | 30 | 30 |
| 11 | Varón | 1 | Axila | 40 | 25 | 35 |
| 12 | Mujer | 14 | Mano | 25 | 70 | 5 |
| 13 | Mujer | 0,4 | Espalda | 50 | 10 | 40 |
| 14 | Varón | 0,75 | MS | 30 | 50 | 10 |
| 15 | Varón | 1 | MS | 30 | 40 | 30 |
| 16 | Varón | 1 | Axila | 40 | 35 | 25 |
| 17 | Mujer | 1 | MS | 30 | 30 | 40 |
| 18 | Varón | 0,75 | Espalda | 35 | 60 | 5 |
| 19 | Mujer | 0,92 | Espalda | 50 | 25 | 25 |
| 20 | Varón | 1 | Perianal | 25 | 25 | 50 |
| 21 | Varón | 5 | Cervical posterior | 50 | 40 | 10 |
| TOTAL | 13:8 | 3,8 | Predominancia de los componentes n (%) | 4 (14%) | 9 (38%) | 1 (0,04%) |
| 7 (33%) | ||||||
HFI: hamartoma fibroso de la infancia; MS: miembro superior.
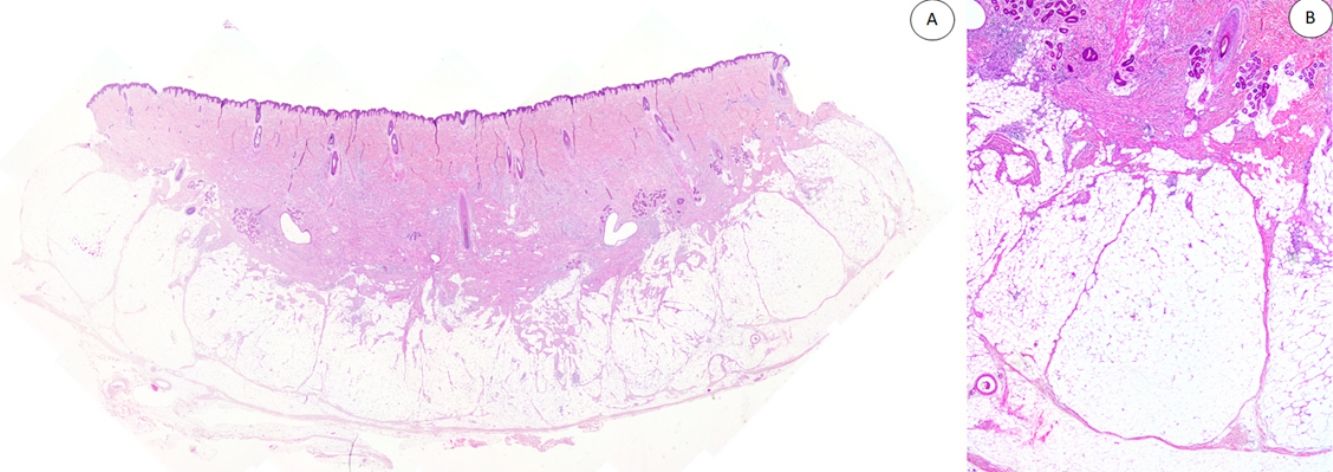
Los casos se caracterizaban por presentar una morfología trifásica con una proporción variable de tejido fibroblástico/miofibroblástico, mesenquimal inmaduro y adiposo maduro en dermis o subcutis (figs. 1, 2 y 3). En términos generales, las lesiones se describieron como mal delimitadas (fig. 1).
 Panorámica de HE que muestra una lesión mal delimitada en dermis reticular con cantidades variables de tejido adiposo, fibroblástico y mesenquimal inmaduro. B) A mayor aumento se observan bandas densas de tejido fibroso proyectándose en el tejido graso maduro. HE: hematoxilina-eosina.")
 HFI dérmico e hipodérmico B) A mayor aumento, bandas densas de tejido fibroso proyectándose en el tejido graso maduro. C) Trabéculas entrelazadas de tejido fibroso que presentan colecciones de fibrocitos de aspecto inmaduro en patrones de espiral. D) Componente mesenquimatoso inmaduro basófilo entremezclado con tejido fibroso. HFI: hamartoma fibroso de la infancia.")
A) HFI dérmico e hipodérmico B) A mayor aumento, bandas densas de tejido fibroso proyectándose en el tejido graso maduro. C) Trabéculas entrelazadas de tejido fibroso que presentan colecciones de fibrocitos de aspecto inmaduro en patrones de espiral. D) Componente mesenquimatoso inmaduro basófilo entremezclado con tejido fibroso.
HFI: hamartoma fibroso de la infancia.
 Panorámica de HF. B, C y D) A mayor aumento, haces y espirales de tejido fibroblástico y mesenquimal inmaduro. HF: hamartoma fibroso.")
Las trabéculas fibrosas varían en grosor y disposición, y contienen haces de células en forma de huso dispersos aleatoriamente entre haces de colágeno (con colecciones de fibroblastos de aspecto inmaduro, formando patrones en espiral) (fig. 2). Los nódulos mixoides de tejido mesenquimal inmaduro estaban compuestos por células redondas o estrelladas primitivas, indiferenciadas, dispuestas libremente o embebidas en un estroma mixoide (fig. 3). Finalmente, podemos observar áreas de tejido adiposo maduro (figs. 1 y 2).
La proporción relativa de cada componente era muy variable. Uno de ellos se consideró «predominante» si ocupaba igual o más del 45% de toda la lesión. Siete casos presentaron una distribución equitativa; en nueve, el componente predominante fue el adiposo, y en cuatro, fue el fibroblástico. Encontramos un predominio significativo del tejido mesenquimal inmaduro en uno de nuestros pacientes (tabla 1).
Se puede observar mínimos focos de infiltrado inflamatorio en cuatro casos y en uno, alguna figura mitótica, pero sin necrosis o atipia nuclear. Ninguno de nuestros pacientes mostró características morfológicas sarcomatosas.
En nuestro estudio, los casos testados para desmina2, podoplanina3, cúmulo de diferenciación 31 (CD31)4, antígeno epitelial de membrana (EMA)3, CD63 (NKI/C3) (Mouse Monoclonal Antibody; anticuerpo monoclonal de ratón contra CD63)2 y la expresión nuclear de beta-catenina nuclear1 fueron negativos (fig. 4).
Discusión Panorámica de una extirpación simple de un HFI subcutáneo. B) A mayor aumento, tejido graso y fibroblástico. C) La actina del músculo liso decora el tejido fibroso maduro. D) Áreas de positividad para CD34, marcando componente inmaduro. HFI: hamartoma fibroso de la infancia.")
En este trabajo describimos una serie de 21 casos de esta entidad poco frecuente. Con una cierta predominancia masculina 1,6:1, es una neoplasia de aparición en la infancia. Si bien, hasta el 20% de los HFI se reconocen al nacimiento, la mayoría se diagnostican alrededor de los dos años de edad9. Saab et al., identificaron el 88% de los casos antes de los dos años, el 7% entre los tres y los cinco años y el 5% después de los cinco años21. En nuestra serie, el 76% de los casos se observaron en lactantes y el 24% tras los dos años de edad. Ocasionalmente, se han descrito en la literatura pacientes diagnosticados entre la segunda, tercera y cuarta década de la vida, probablemente debido a un retraso en el diagnóstico y no a una verdadera presentación tardía de la entidad. Ji et al., publicaron un paciente diagnosticado a los 47 años de edad pese a presentar una lesión de lento crecimiento progresivo en la región craneocervical izquierda de 42 años de evolución27. De forma similar, en nuestra serie presentamos un paciente de 24 años que reconoció la aparición de la lesión en la infancia.
En cuanto a la localización, nuestros casos tienen predilección por el miembro superior (n = 8, 40%), la espalda (n = 5, 24%) y la axila (n = 3). Los estudios iniciales del HFI enfatizaban dichas ubicaciones como sitios de preferencia, si bien con el tiempo otras han sido publicadas. Pese a que hasta el 20% de las lesiones en la literatura aparecen en área genital, en nuestros casos solo había una en la región perianal9,20–22.
La variabilidad clínica y baja frecuencia de esta neoplasia benigna, que puede tener patrón infiltrativo, plantea como problema principal el diagnóstico diferencial con otras entidades que requieren una actitud más agresiva. El correcto diagnóstico depende, en gran medida, de la identificación morfológica de los tres componentes histológicos9,12,16,17,28–30. En nuestra opinión, es importante localizar el componente mesenquimal inmaduro que, independientemente de su proporción, siempre debe estar presente. Una histología trifásica (tejido mesenquimal inmaduro, fibroblástico y adiposo) nos permitir diferenciar tumoraciones con predominio de tejido adiposo como: la lipofibromatosis, el tumor neural similar a la lipofibromatosis (donde encontramos además cierta atipia citológica) y el lipoblastoma; de predominio fibroblástico como: la fibromatosis de tipo desmoide (poco frecuente en niños y proliferación compacta de miofibroblastos), los miofibromas (patrón bifásico, puede presentar necrosis y/o calcificación hacia el centro de la lesión), los fibromas aponeuróticos calcificantes (que contienen además nódulos calcificados rodeados de células epitelioides), el nevus fibroblástico del tejido conectivo (puede reconocerse por la localización dérmica y la ausencia de mesénquima inmaduro) y el fibroblastoma de células gigantes. Las hendiduras presentes en el fibroblastoma pueden estar vacías u ocupadas por material amorfo mucinoso y están revestidas por una hilera discontinua de células tumorales. En este tumor, además, es frecuente encontrar células multinucleadas y presenta reordenamiento de la subunidad B del factor de crecimiento derivado de plaquetas (PDGFB)1,9,21,24,28. Aunque ambas entidades tienen hendiduras, las zonas hialinizadas con grietas del HFI, también denominadas patrón pseudoangiomatoso, habitualmente son fácilmente distinguibles por patólogos experimentados. Respecto a este último patrón en HFI, su frecuencia es variable entre series (hasta en un 53% de los HFI del estudio de Saab et al.21 y hasta en un 30% en el de Abrahami et al.9; frente al 14% encontrado en nuestra serie), es característicamente negativo para marcadores endoteliales más específicos que CD34, como CD31 y podoplanina (D2-40), puesto que sus hendiduras no están tapizadas por endotelio1,9,21,24,28. En caso del predominio de componente mesenquimal inmaduro, además de por las características propias de cada tumor, podemos distinguir el schwannoma celular, el tumor maligno de la vaina del nervio periférico y el neurofibroma (monomorfo) por la fuerte positividad para S100 en el tejido mesenquimal, a diferencia del HFI donde solo se tiñe el tejido adiposo28–30.
En conclusión, el HFI es considerado un proceso neoplásico benigno de la infancia con una histología trifásica característica cuyo tratamiento de elección es la escisión quirúrgica. Consideramos prioritario identificar su componente mesenquimal inmaduro para facilitar el diagnóstico histopatológico. Nuestros resultados confirman los hallazgos clínicos, histológicos e inmunohistoquímicos observados previamente en la literatura y son ejemplos iconográficos que esperamos que puedan ayudar a realizar un diagnóstico diferencial con otras entidades, evitando así conductas agresivas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


