




Fibrous hamartoma of infancy (FHI) is a rare soft-tissue tumor usually seen in infants and young children. Histologically, the tumor is characterized by a triphasic morphology, and the clinical course is benign.
MethodsWe described the histopathologic and immunohistochemical features in a series of 21 cases of FHI and reviewed the main entities to consider in differential diagnosis.
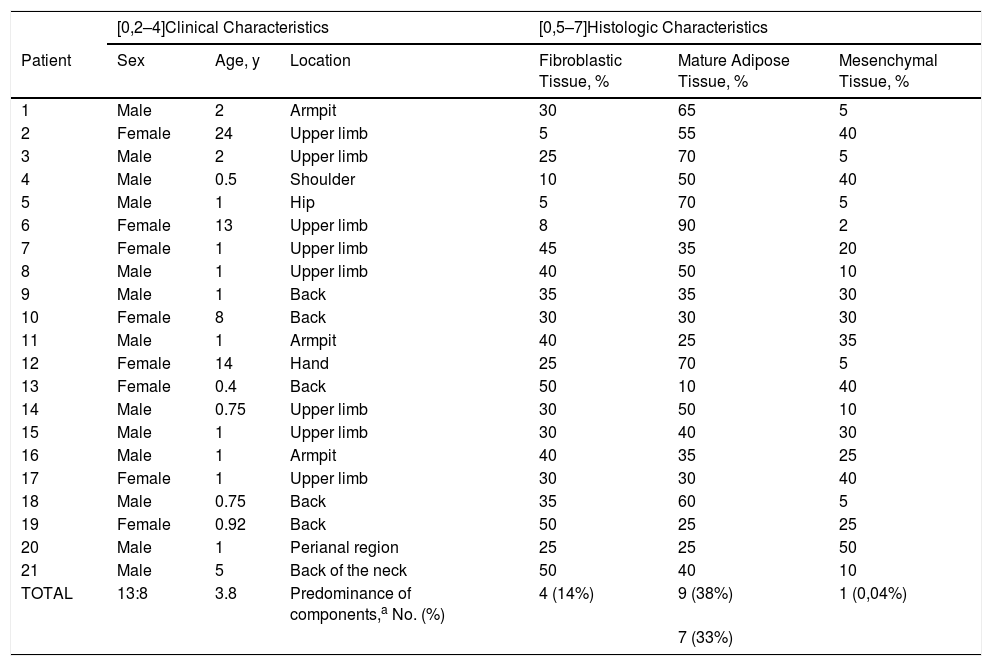
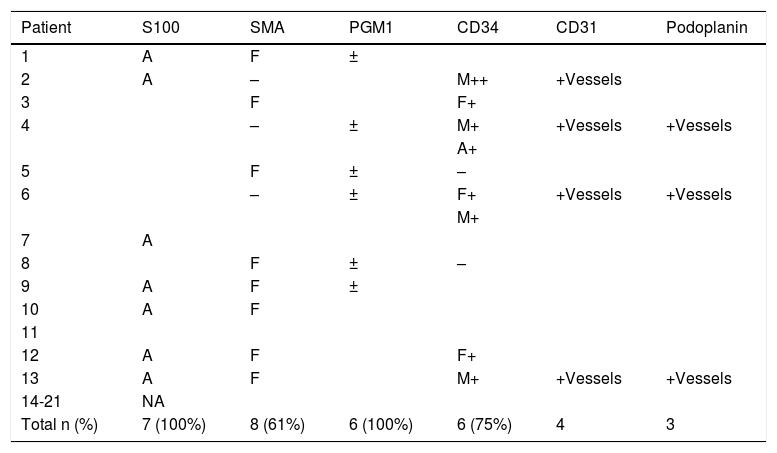
ResultsMost patients in the series were male. The mean age was 3.7 years (range, 5 months–24 years), and lesions were found in locations that have been previously reported. Histopathologically, the tumors were composed of variable proportions of fibroblastic, mesenchymal, and mature adipose tissue. Three of the 13 immunohistochemically stained biopsies (14%) contained hyalinized zones with cracking artifacts, mimicking giant cell fibroblastoma. Eight of the 13 stained with smooth-muscle actin (61%) were positive in the fibroblastic component, 6 of the 8 stained with CD34 (75%) were positive in the immature mesenchymal and fibroblastic components, and all 7 stained with S100 were positive (100%) in adipose tissue.
ConclusionsOur histopathologic findings are consistent with those described in larger series. However, in order to reach a precise diagnosis and plan treatment, clinical heterogeneity obliges us to become familiar with variations in the characteristic triphasic histology of FHI.
El Hamartoma fibroso de la infancia (HFI) es tumor de tejidos blandos poco frecuente de lactantes y niños, caracterizado histopatólogicamente por su morfología trifásica y curso benigno.
MétodosPresentamos las características histopatológicas e inmunohistoquímicas de una serie de 21 casos de HFI y revisamos sus principales diagnósticos diferenciales.
ResultadosLas lesiones predominaron en varones con una edad media de 3,7 años (5 meses-24 años) en localizaciones previamente descritas. Histopatológicamente, se caracterizaron porproporciones variables de tejido fibroblástico, mesenquimal y adiposo maduro. Tres casos (21%) mostraron zonas hialinizadas con artefactos en grietas, que imitaban al fibroblastoma de célulasgigantes. Presentaron tinción positiva para actina de músculo liso 8/13 (61%) en el componentefibroblástico, CD34 en 6/8 (75%) en el mesenquimal inmaduro y fibroblástico; y S100 en el tejidoadiposo en 7/7 (100%).
ConclusiónNuestros hallazgos histopatológicos son superponibles a los descritos en series amplias. No obstante, la heterogeneidad clínica obliga a conocer las variantes de esta entidad dentro de su típica histomorfología trifásica, para lograr un correcto diagnóstico y manejo terapéutico.
The World Health Organization (WHO) has defined fibrous hamartoma of infancy (FHI) as a pediatric, benign, poorly delimited tumor with a characteristic triphasic histologic appearance comprising fibroblastic fascicles, mature adipose tissue, and clusters of immature mesenchymal cells in a primitive myxoid stroma.1 FHI was first described in 1956 by Reye2 as a reactive phenomenon, which he called subdermal fibromatous tumor of infancy. Based on its clinical presentation and hamartomatous features, it was renamed fibrous hamartoma of infancy in a later case report by Enzinger.3 In 2005, isolated reports of complex cytogenetic alterations—t (6; 12; 8) (q25; q24.3; q13), t (2; 3) (q31; q21)—and chromosomal rearrangements (1, 2, 4 y 17)4–6 in patients with FHI started to appear. It was not until 2016, however, that Park et al.7 described an epidermal growth factor receptor (EGFR) exon 20 insertion/duplication mutation in 12 patients with FHI, suggesting that the condition might be neoplastic. Ellington et al.8 subsequently reported another case involving the same mutation.8 In a genomic microarray analysis of 8 cases of FHI, Al-Ibraheemi et al.9 detected genomic copy number changes in the 2 cases with sarcomatous features on histology. Both patients had a complex molecular karyotype (hyperdiploid/tetraploid) involving a loss of heterozygosity of chromosomes 1 p and 11p in one case and a loss of chromosome 14 and fragments 10p and 22q11 in the other. Identification of the same cytogenetic alterations in different components of the lesion supports the idea that FHI is a neoplastic process. We agree with this consideration.
Although there are no known associations between FHI and family disorders or syndromes, in our review of the literature, we found one report of tuberous sclerosis10 and another of Williams syndrome11 associated with FHI. In the second case, the authors observed minimal elastin expression in the tumor and hypothesized that a microdeletion in the elastin gene might predispose to the development of FHI.
FHI is clinically heterogeneous and can affect a wide variety of locations. It typically presents as a solitary, poorly delimited, nodule measuring approximately 0.5 to 10 cm in diameter that does not affect the epidermis.12 Other clinical variants, however, have been described and include multicentric13,14 and giant lesions (up to 20 cm)15 and lesions with hyperpigmentation,16 hypertrichosis, and hyperhidrosis.17–19
FHI lesions are usually asymptomatic, but they can present as infiltrating nodules or nodules fixed to the deep planes, making them difficult to distinguish from malignant tumors.20 They mostly occur in the axilla, upper back, arms, and genital area.9,21,22 They grow rapidly in the first 5 years of life and then stabilize. No cases of involution or metastasis have been described, but there have been 2 reports of tumors with sarcomatous areas. The treatment of choice is complete surgical excision with clear margins.22 Recurrence rates lie around 15%,15,16,23–26 although rates as low as 1% have been reported by specialized centers.9
Immunohistochemical findings in FHI are nonspecific and similar to those observed in other tumors. The fibroblastic component is typically positive for vimentin and smooth-muscle actin, but it may also show focal reactivity to CD68, desmin, and factor XIIIa. Unlike the adipose component, it is negative for S100 proteint. The immature component is typically positive for CD34.21,25
In this article, we describe the main histopathologic and immunohistochemical features of 21 cases of FHI and review the main entities that should be considered in the differential diagnosis according to the predominant component observed.
Materials and MethodsWe performed a retrospective observational study of FHI cases diagnosed between 1990 and 2015 at the Voth Medical Center in Madrid, Spain; the Dermopathology Department at Hospital Universitario de la Princesa in Madrid; and the Friedrichshafen Dermatopathologie center in Friedrichshafen, Germany. In all cases, the tissue specimens were fixed in 5% buffered formalin, routinely processed, and embedded in paraffin. The sections for histopathologic examination (4 µm) were stained with hematoxylin-eosin, while the immunohistochemical sections (also 4 μm) were mounted on positively charged slides. These were left to dry overnight at 45 °C. The slides were deparaffinized in xylol for 30 minutes, rehydrated in a descending series of ethanol, and incubated in EDTA for 30 minutes at 95 °C (pH = 9.0). They were then cooled at room temperature for 20 minutes. After rapid cooling with alkaline phosphatase and biotin blocking agents, avidin was used to incubate the sections with the different antibodies used (Table 2).
Clinical and Histologic Characteristics of Patients With Fibrous Hamartoma of Infancy.
| [0,2–4]Clinical Characteristics | [0,5–7]Histologic Characteristics | |||||
|---|---|---|---|---|---|---|
| Patient | Sex | Age, y | Location | Fibroblastic Tissue, % | Mature Adipose Tissue, % | Mesenchymal Tissue, % |
| 1 | Male | 2 | Armpit | 30 | 65 | 5 |
| 2 | Female | 24 | Upper limb | 5 | 55 | 40 |
| 3 | Male | 2 | Upper limb | 25 | 70 | 5 |
| 4 | Male | 0.5 | Shoulder | 10 | 50 | 40 |
| 5 | Male | 1 | Hip | 5 | 70 | 5 |
| 6 | Female | 13 | Upper limb | 8 | 90 | 2 |
| 7 | Female | 1 | Upper limb | 45 | 35 | 20 |
| 8 | Male | 1 | Upper limb | 40 | 50 | 10 |
| 9 | Male | 1 | Back | 35 | 35 | 30 |
| 10 | Female | 8 | Back | 30 | 30 | 30 |
| 11 | Male | 1 | Armpit | 40 | 25 | 35 |
| 12 | Female | 14 | Hand | 25 | 70 | 5 |
| 13 | Female | 0.4 | Back | 50 | 10 | 40 |
| 14 | Male | 0.75 | Upper limb | 30 | 50 | 10 |
| 15 | Male | 1 | Upper limb | 30 | 40 | 30 |
| 16 | Male | 1 | Armpit | 40 | 35 | 25 |
| 17 | Female | 1 | Upper limb | 30 | 30 | 40 |
| 18 | Male | 0.75 | Back | 35 | 60 | 5 |
| 19 | Female | 0.92 | Back | 50 | 25 | 25 |
| 20 | Male | 1 | Perianal region | 25 | 25 | 50 |
| 21 | Male | 5 | Back of the neck | 50 | 40 | 10 |
| TOTAL | 13:8 | 3.8 | Predominance of components,a No. (%) | 4 (14%) | 9 (38%) | 1 (0,04%) |
| 7 (33%) | ||||||
Immunohistochemical Findings.a
| Patient | S100 | SMA | PGM1 | CD34 | CD31 | Podoplanin |
|---|---|---|---|---|---|---|
| 1 | A | F | ± | |||
| 2 | A | – | M++ | +Vessels | ||
| 3 | F | F+ | ||||
| 4 | – | ± | M+ | +Vessels | +Vessels | |
| A+ | ||||||
| 5 | F | ± | – | |||
| 6 | – | ± | F+ | +Vessels | +Vessels | |
| M+ | ||||||
| 7 | A | |||||
| 8 | F | ± | – | |||
| 9 | A | F | ± | |||
| 10 | A | F | ||||
| 11 | ||||||
| 12 | A | F | F+ | |||
| 13 | A | F | M+ | +Vessels | +Vessels | |
| 14-21 | NA | |||||
| Total n (%) | 7 (100%) | 8 (61%) | 6 (100%) | 6 (75%) | 4 | 3 |
Abbreviations: NA, nonapplicable; S100, S100 protein; SMA, smooth-muscle actin.
Positivity for a given tissue is shown with the corresponding letter (A, adipose; F, fibroblastic; M, mesenchymal). Stains that were not predominant for a given tissue are marked as + (positive), – (negative), or ± (weak). Samples not stained with a given antibody are not shown. Samples tested for desmin (n = 2), NKIC3 (n = 2), epithelial membrane antigen (n = 3), and nuclear β-catenin expression (n = 1) are not included.
This study was approved by a research ethics committee and conducted in accordance with the principles of the Declaration of Helsinki.
ResultsClinical and Epidemiological CharacteristicsThe tissue specimens were from 13 males and 8 females with a median age at diagnosis of 1 year (range, 5 months to 24 years). Eighteen of the lesions were located in typically described sites (axilla, upper limb, dorsal region, and back of the neck). The other 3 were located in less usual sites (hip, hand, and perianal region) (Table 1).
Histopathologic FindingsAll the lesions had the characteristic triphasic histologic appearance with variable proportions of fibroblastic/myofibroblastic tissue, immature mesenchymal tissue, and mature adipose tissue in the dermis or subcutaneous tissue (Figs. 1–3). The lesions were largely described as poorly delimited (Fig. 1).
 showing a poorly delimited lesion in the reticular dermis with variably sized adipose, fibroblastic, and immature mesenchymal tissue components. B, Higher-magnification view showing dense bands of fibrous tissue projecting into the mature adipose tissue.")
A, Panoramic view (hematoxylin-eosin) showing a poorly delimited lesion in the reticular dermis with variably sized adipose, fibroblastic, and immature mesenchymal tissue components. B, Higher-magnification view showing dense bands of fibrous tissue projecting into the mature adipose tissue.

A, Fibrous hamartoma of infancy in the dermis and subcutaneous tissue. B, Higher-magnification view showing dense bands of fibrous tissue projecting into the adipose tissue. C, Interlacing trabeculae of fibrous tissue containing clusters of immature fibrocytes forming whorls. D, Immature basophilic mesenchymal component interspersed with fibrous tissue.
 of a fibrous hamartoma. B, C, and D, Higher-magnification view showing bundles and whorls of fibroblastic and immature mesenchymal tissue.")
The fibrous trabeculae varied in thickness and distribution and contained bundles of spindle cells randomly scattered among the collagen bundles (with clusters of immature fibroblasts, forming whorls) (Fig. 2). The myxoid nodules of immature mesenchymal tissue were composed of undifferentiated primitive round or stellate cells arranged freely or embedded in a myxoid stroma (Fig. 3). Areas of mature adipose tissue were also visible (Figs. 1 and 2).
The relative proportion of the different tissue components was highly variable. A component was considered to be predominant if it occupied 45% or more of the entire lesion. Seven lesions had no predominant component and 9 had a predominant adipose component, 4 a predominant fibroblastic component, and 1 a significantly predominant immature component (Table 1).
Examination showed small foci of inflammatory infiltrates in 4 lesions and mitotic figures in another. There were no signs of necrosis or nuclear atypia. None of the tumors had sarcomatous features.
The specimens tested with desmin,2 podoplanin,3 CD31,4 epithelial membrane antigen,3 CD63 (NKI/C3) mouse monoclonal antibody,2 and nuclear β-catenin all showed negative results (Fig. 4).
Discussion
We have described a series of 21 cases of FHI, a rare tumor. FHI is slightly more common in males (male to female ratio, 1.6:1) and appears in infancy. Around 20% of cases are detected at birth, but most are diagnosed around the age of 2 years.9 In the series by Saab et al.,21 88% of FHI cases were detected before the age of 2 years, 7% between the ages of 3 and 5 years, and just 5% at an older age. In our series, 76% of cases were diagnosed in infants younger than 2 years. There have been some reports of FHI being diagnosed in the second, third, and fourth decades of life, but they probably correspond to a late diagnosis rather than a late age of onset. Ji et al.27 described the case of a patient who was diagnosed with FHI at 47 years of age, even though he had had a slow-growing lesion in the left craniocervical region for 42 years. Similarly, the 24-year-old patient diagnosed with FHI in our series reported that the lesion had been present since childhood.
The most common location for FHI in our series was the upper limb (n = 8, 40%), followed by the back (n = 5, 24%), and the axilla (n = 3). Early studies of FHI described these sites as preferential locations, but with time, new locations have been reported. Approximately 20% of FHI lesions described in the literature occur in the perianal region,9,20–22 but in our series, just 1 patient had a lesion in this area.
The clinical variability and low prevalence of FHI are the main reasons why it is difficult to distinguish this benign tumor, which can sometimes exhibit infiltrative growth, from other tumors requiring more aggressive treatment. Accurate diagnosis of FHI largely depends on the identification of the characteristic triphasic morphology.9,12,16,17,28–30 In our opinion, detection of the immature mesenchymal component is key, as this is always present to a greater or lesser extent. Observation of the triphasic component (immature mesenchymal, fibroblastic, and adipose tissue) during histology will help distinguish FHI from tumors with a predominant adipose tissue component—such as lipofibromatosis, lipofibromatosis-like neural tumor (which also displays some cytologic atypia), and lipoblastoma—and tumors with a predominant fibroblastic component—such as desmoid-type fibromatosis (rare in children and characterized by a dense proliferation of myofibroblasts), myofibroma (biphasic pattern and possibly necrosis and/or calcification towards the center of the lesion), calcifying aponeurotic fibroma (which also contains calcified nodules surrounded by epithelioid cells), fibroblastic connective tissue nevus (dermal location and absence of immature mesenchymal tissue), and giant cell fibroblastoma. Cleft-like spaces in fibroblastoma may be empty or occupied by amorphous mucinous material and they are lined with a discontinuous row of tumor cells. Other findings are frequent multinucleated cells and platelet-derived growth factor PDGFB subunit β rearrangement.1,9,21,24,28 Although both FHI and fibroblastoma have clefts, the characteristic pseudoangiomatous pattern of FHI, formed by hyalinized areas with slit-like spaces, is normally easy to spot for an experienced pathologist. The prevalence of this pseudoangiomatous pattern varies (14% in our series compared with 53% in the series by Saab et al.21 and 30% in the series by Abrahimi et al.9). Staining for endothelial markers with a higher specificity than CD34, such as CD31 and podoplanin (D2-40), is normally negative since the cleft-like spaces are not lined with endothelial cells.1,9,21,24,28 FHI with a predominant immature mesenchymal tissue component must be distinguished from cellular schwannoma, malignant peripheral nerve sheath tumor, and neurofibroma (monomorphous). These tumors will show strong positivity for S100 protein in the mesenchymal tissue, unlike FHI, where S100 protein is only expressed in the adipose tissue component.28–30
In conclusion, FHI is a benign tumor of infancy with a characteristic triphasic histologic appearance. The treatment of choice is surgical excision. Identification of an immature mesenchymal component is key to histopathologic diagnosis. The clinical, histologic, and immunohistochemical findings in this series of 21 patients with FHI are consistent with previous reports and provide important visual clues to aid the differential diagnosis and avoid unnecessary aggressive treatment.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Martos-Cabrera L, Sampedro-Ruiz R, Pérez-González YC, Mentzel T, Llamas-Velasco M. Hamartoma fibroso de la infancia: una serie de 21 casos y revisión de la literatura. Actas Dermosifiliogr. 2021;112:520–527.


