



El granuloma anular (GA) es una dermatosis de etiología desconocida caracterizada histopatológicamente por 3 patrones: 1) granulomas necrobióticos en dermis media-superficial con una o más áreas de necrobiosis con aumento de mucina, rodeadas de histiocitos y linfocitos; 2) la variante intersticial o incompleta con aumento de linfocitos e histiocitos entre los haces de colágeno que están separados por mucina, y 3) la presencia de granulomas tuberculoides o sarcoideos1. Se han descrito variantes clínicas que incluyen la forma localizada, la generalizada, la perforante y la subcutánea, así como otras aún más raras como son la palmo-plantar o la de tipo parche2. El GA localizado es el prototipo de la enfermedad y se caracteriza por pápulas y placas de tonalidad rosa o rojiza, con conformación anular, sin componente epidérmico y habitualmente localizado en las extremidades. A nivel histopatológico, se pueden destacar las variantes elastolíticas pero el GA también puede asociar siringometaplasia escamosa ecrina o elastólisis de la dermis media e incluso presencia de vasculitis y neutrófilos2. Más recientemente se ha descrito el GA pseudolinfomatoso1.
Presentamos un caso de GA histológicamente peculiar, la variante pseudolinfomatosa.
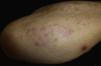
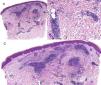
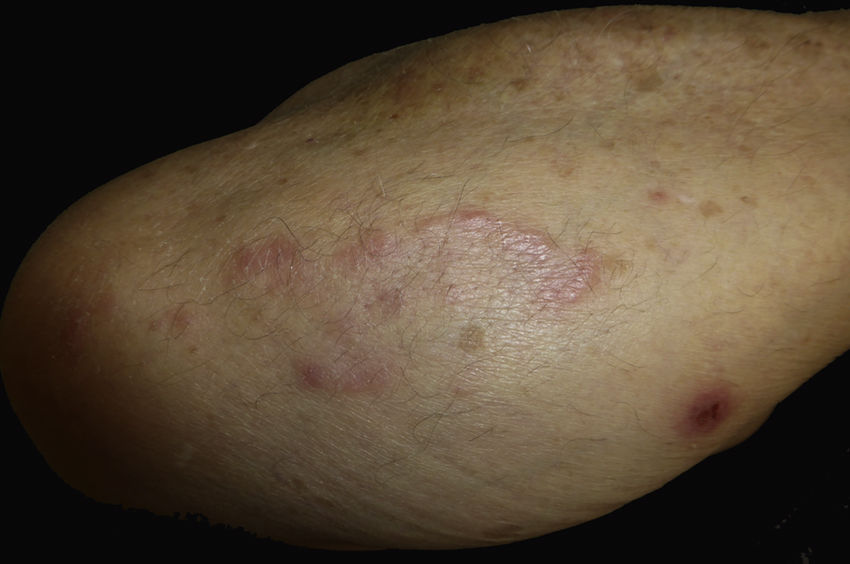
Caso clínico. Varón de 82 años con antecedentes de hipertensión, miocardiopatía dilatada y neoplasia maligna de próstata que acude por la aparición de una lesión pruriginosa localizada en el dorso del antebrazo derecho de un año de evolución. A la exploración se observaban pápulas eritematosas confluyendo para formar una placa de aspecto arciforme, no infiltrada, de 5×2cm (fig. 1). La biopsia mostró un infiltrado linfocítico e histiocítico intersticial acompañado por un infiltrado linfocítico perivascular prominente sin presencia de células atípicas. La tinción azul alcián permitió demostrar un aumento de mucina intersticial (fig. 2a-c y fig. 3a). Inmunohistoquímicamente, el infiltrado perivascular estaba formado por una mezcla de linfocitos CD4 y CD8 con numerosas células CD163 y CD68 positivas entre los haces de colágeno (fig. 3b-d). No se observaron grupos de células dendríticas plasmacitoides con CD123. Con estos datos clínicos e histopatológicos se realizó el diagnóstico de GA pseudolinfomatoso. La lesión se resolvió por completo tras un mes de aplicación tópica diaria de propionato de clobetasol al 0,05% en crema, sin recurrencias hasta el momento actual.
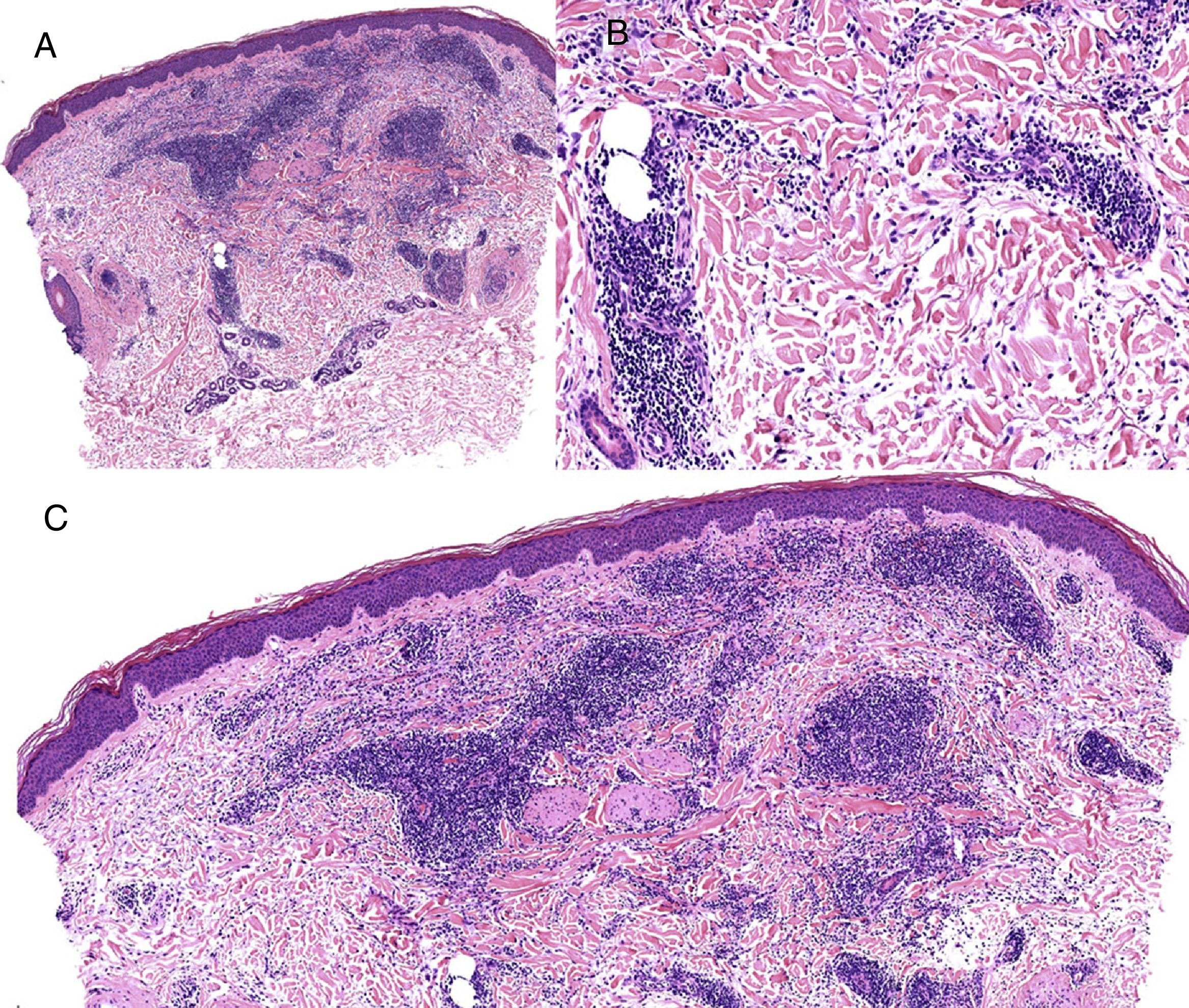
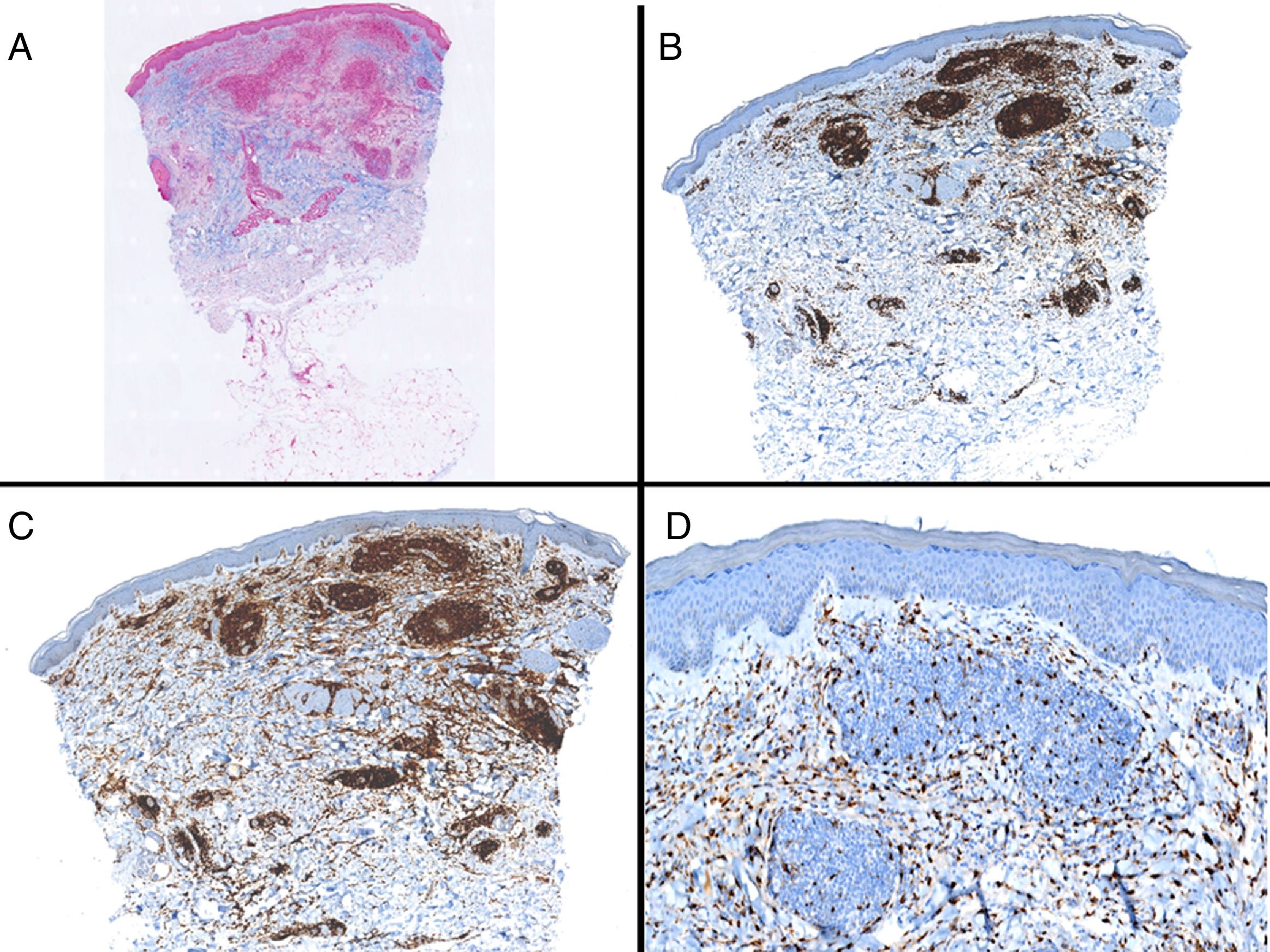
A) Panorámica de la lesión con azul alcián. B) Panorámica mostrando la distribución principalmente perivascular de los linfocitos CD3 positivos. C) Panorámica mostrando que la mayoría de los linfocitos T presentes son CD4. D) Tinción con CD68 mostrando la presencia de macrófagos intersticiales.
El GA pseudolinfomatoso, descrito por Cota et al. en el año 2012, se caracteriza histopatológicamente por la presencia de un denso infiltrado linfocítico alrededor de los vasos superficiales y profundos; por la ausencia de linfocitos atípicos; y por la presencia concomitante de GA intersticial o necrobiótico1. Clínicamente, en la serie de Cota et al., se plantearon diagnósticos muy variados (pseudolinfomas, micosis, dermatitis liquenoide, sarcoidosis, dermatitis papular, parapsoriasis en placas o eritema figurado) y solo 3 de los casos fueron remitidos con diagnóstico clínico inicial de GA. El 60% de sus pacientes mostraban lesiones localizadas, como la observada en nuestro caso.
Desde un punto de vista histopatológico, en el diagnóstico diferencial del GA pseudolinfomatoso se incluyen la hiperplasia linfoide, el lupus túmido (que puede ser excluido por la clínica y por el patrón intersticial presente en la biopsia), la micosis fungoide intersticial (variante histopatológica de micosis fungoide caracterizada por la presencia de linfocitos dispersos entre las fibras de colágeno y en el que nunca hay más macrófagos intersticiales que linfocitos) y la reacción medicamentosa granulomatosa intersticial, que en nuestro caso podría ser excluida por anamnesis.
Se han descrito infiltrados granulomatosos, que pueden aparecer junto con las células atípicas tumorales específicas, tanto en linfomas de Hodgkin, no Hodgkin y en algunos tumores sólidos3,4. En nuestro paciente, la rápida resolución, tal y como está descrita en el GA localizado, la clínica típica de GA y la ausencia de otras patologías, permite excluir esta posibilidad con un alto grado de certeza. La relación entre GA y tumores malignos es probablemente casual5.
Se han publicado casos aislados de coexistencia de GA con procesos linfoides como leucemia/linfoma T del adulto3,6, leucemia aguda mieloide o linfoma primario cutáneos T CD4 de célula pequeña-mediana4. Dentro de este segundo grupo, que podríamos considerar como manifestaciones no específicas de linfomas, también pueden englobarse los casos de sarcoidosis y linfoma.
Por tanto, el diagnóstico de GA pseudolinfomatoso debe basarse en la correlación clínico-patológica y debe apoyarse en los correspondientes estudios inmunohistoquímicos para descartar linfoma u otras neoplasias concomitantes, sobre todo si se visualizan células atípicas, y en la realización de serologías o PCR de Borrelia para descartar borreliosis, en áreas endémicas o ante cuadros clínicos compatibles.
En casos histopatológicamente caracterizados por GA intersticial o con presencia de granulomas necrobióticos, la presencia de un denso infiltrado linfoide perivascular superficial y profundo obliga a tener en cuenta la posibilidad de un GA pseudolinfomatoso, entidad rara, escasamente descrita en la literatura, pero que hay que conocer para evitar un sobretratamiento del paciente o la realización de pruebas complementarias innecesarias.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


