







INTRODUCCIÓN
El término fibroxantoma atípico fue acuñado por Helwig en 1961 para definir un tumor poco frecuente con marcado pleomorfismo y con un curso usualmente benigno 1 . Su histogénesis es incierta, pero la mayoría de los autores lo consideran una variante superficial del histiocitoma fibroso maligno y como él derivaría de un progenitor mesenquimatoso pobremente diferenciado que se puede diferenciar hacia línea histiocítica o fibroblástica 2,3 . Generalmente se manifiesta como un nódulo solitario en zonas de daño actínico de personas mayores, aunque con menor frecuencia puede afectar al tronco y a los miembros de personas jóvenes.
El objetivo principal de este trabajo es presentar las características clínicas e histológicas de 10 pacientes recogidos en los últimos 5 años en nuestro hospital.
MATERIAL Y MÉTODOS
Se estudian 10 casos de fibroxantoma atípico diagnosticados en los últimos 5 años en nuestro hospital. Los datos clínicos se recogieron retrospectivamente de la historia clínica, analizándose la edad al diagnóstico, el tiempo desde la aparición de la lesión hasta el diagnóstico, la localización del tumor, los factores predisponentes, enfermedad acompañante y evolución. Los datos histológicos se obtuvieron revisando las preparaciones anatomopatológicas, analizándose el patrón arquitectural, celularidad, tipos celulares presentes, ulceración de las epidermis, invasión vascular o perineural, afectación del tejido celular subcutáneo, grado de pleomorfismo, cantidad de mitosis y presencia de infiltrado inflamatorio acompañante. El índice mitótico fue expresado según el número de mitosis por campo de gran aumento: +corresponde a 3 o menos mitosis por campo; 2 +, entre 4-15 mitosis, y 3 +, más de 15 mitosis por campo.

TABLA 1. DATOS CLÍNICO-EPIDEMIOLÓGICOS DE UNA SERIE DE PACIENTES CON FIBROXANTOMA ATÍPICO
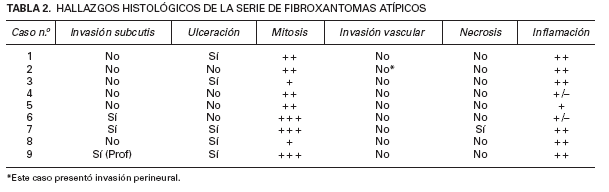
TABLA 2. HALLAZGOS HISTOLÓGICOS DE LA SERIE DE FIBROXANTOMAS ATÍPICOS
A todas las preparaciones se les realizaron las siguientes tinciones de inmunohistoquímica: vimentina (V9), S100, HMB45, panqueratina (AE1/AE3), panactina (HHF-35), desmina (D33), CD68 (KP1) y CD34 (Qbend10).
DESCRIPCIÓN DE LOS CASOS
Las características clínicas se resumen en la tabla 1. Todos los pacientes presentaron lesiones únicas, de diversas morfologías (figs. 1-3), generalmente nodulares, que oscilaban entre 0,5 y 3 cm, algunas erosionadas. Las lesiones se localizaban en 9 casos en cabeza y cuello, mientras que en un caso se localizó en tronco. La edad al diagnóstico osciló entre 25 y 89 años, con una media de 70,8 y una mediana de 74,5 años. El tiempo al diagnóstico presentó una media de 4 meses, con un rango de 2-12 meses.
En todos los casos se realizó extirpación con margen clínico, permaneciendo todos los pacientes vivos y sin lesiones después de un tiempo de evolución que variaba entre 10 y 74 meses, con una media de 39,8 meses y una mediana de 34,5. Sólo un caso presentó recidiva a los 2 meses de la exéresis, realizándose nueva extirpación sin haberse producido recurren-cia 50 meses después. Ningún caso ha desarrollado metástasis locorregionales ni a distancia.
En todos los casos el diagnóstico definitivo se realizó mediante el correspondiente estudio histológico con tinciones de hematoxilina-eosina y posterior inmunohistoquímica. La tabla 2 resume una serie de hallazgos histológicos.
Todos los pacientes presentaban tumores bien circunscritos, no encapsulados, con alta celularidad y centrados en dermis, aunque en 3 casos llegaban hasta el tejido celular subcutáneo. Había un marcado pleomorfismo y polimorfismo. En todas las piezas se observaban tres tipos celulares. La célula predominante era fusocelular, de núcleo grande y vesiculoso, en ocasiones xantomizadas, dispuestas de forma desordenada o en un patrón vagamente estoriforme (figs. 4 y 5). Se acompañaban en mayor o menor grado de células gigantes, algunas multinucleadas y de células poligonales, con citoplasma amplio y eosinófilo (fig. 6).
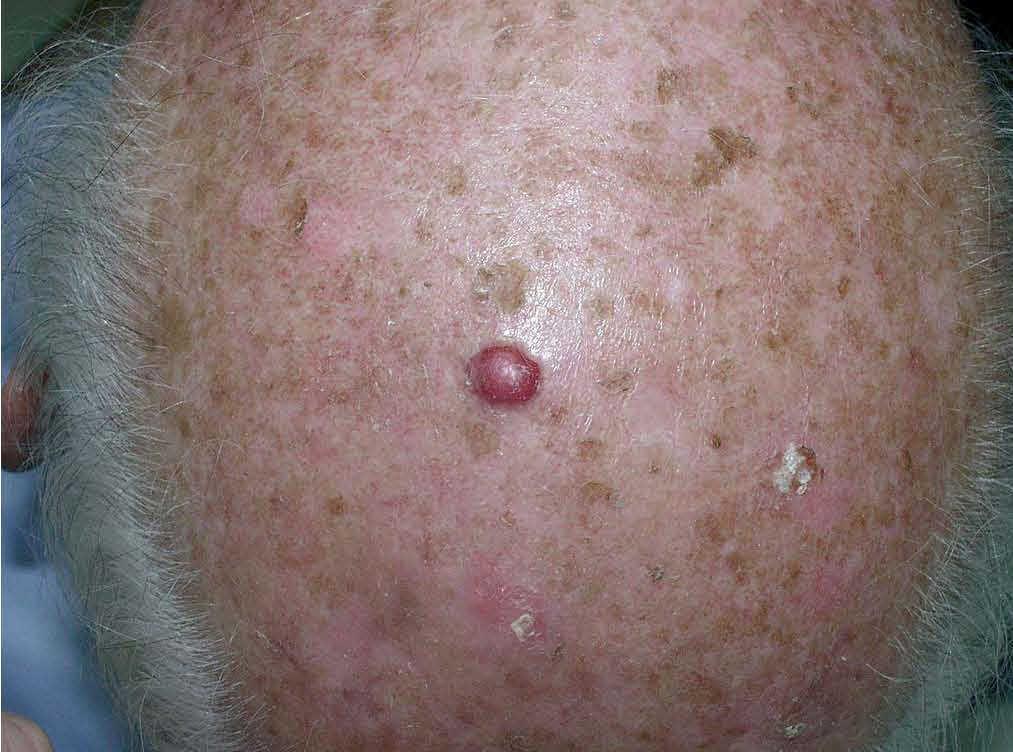
Fig. 1.—Pápula eritematosa en piel de cuero cabelludo con numerosas queratosis actínicas.
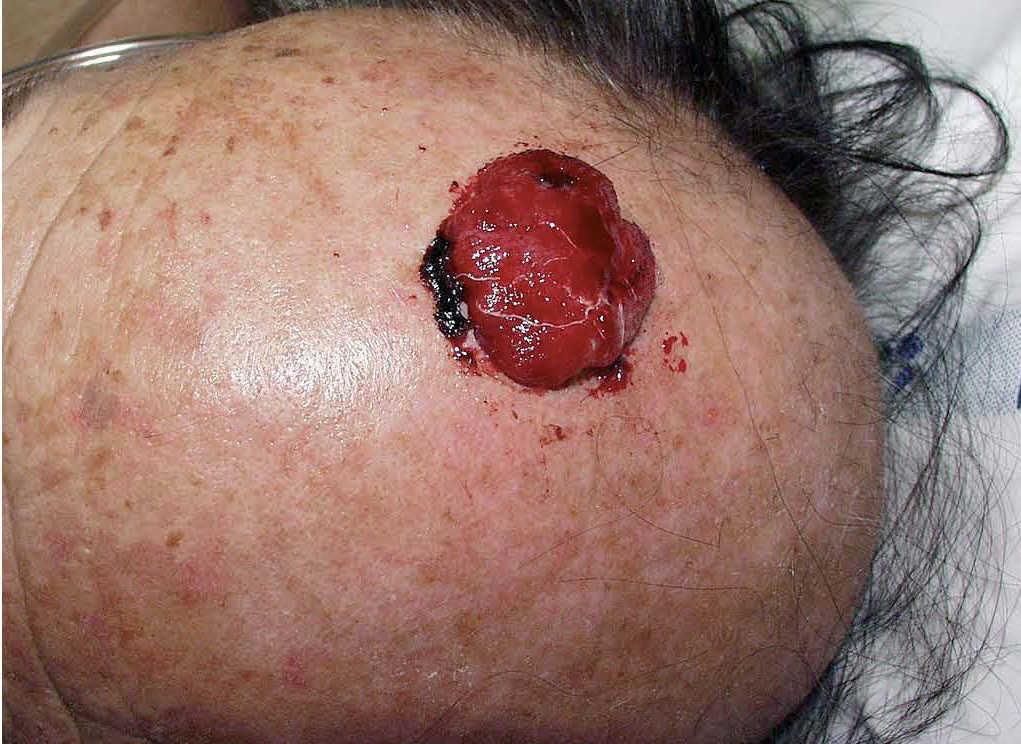
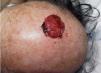
Fig. 2.—Gran masa tumoral, carnosa, friable y sangrante en cuero cabelludo.
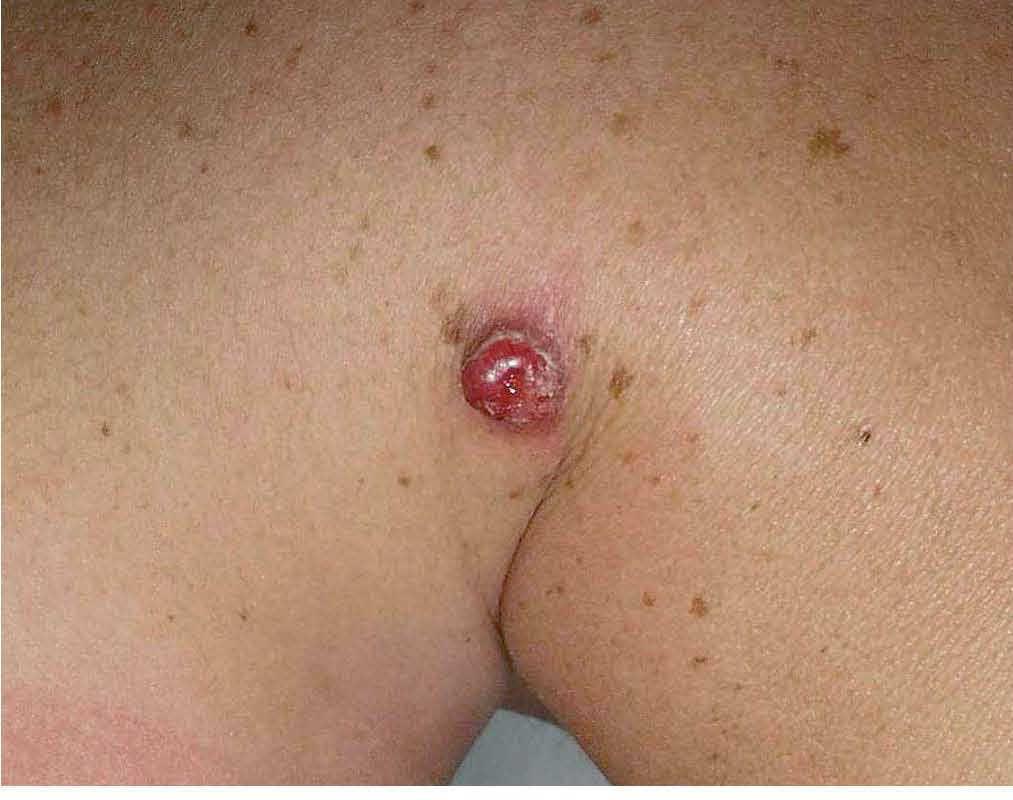
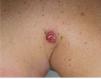
Fig. 3.—Lesión papulosa, angiomatosa, con collarete descamativo en zona del hombro.

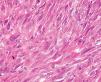
Fig. 4.—El tumor está compuesto predominantemente por células fusiformes pleomórficas, dispuestas en un patrón vagamente estoriforme. (Hematoxilina-eosina, ×100.)

Fig. 5.—Detalle de la anterior. (Hematoxilina-eosina, ×400.)
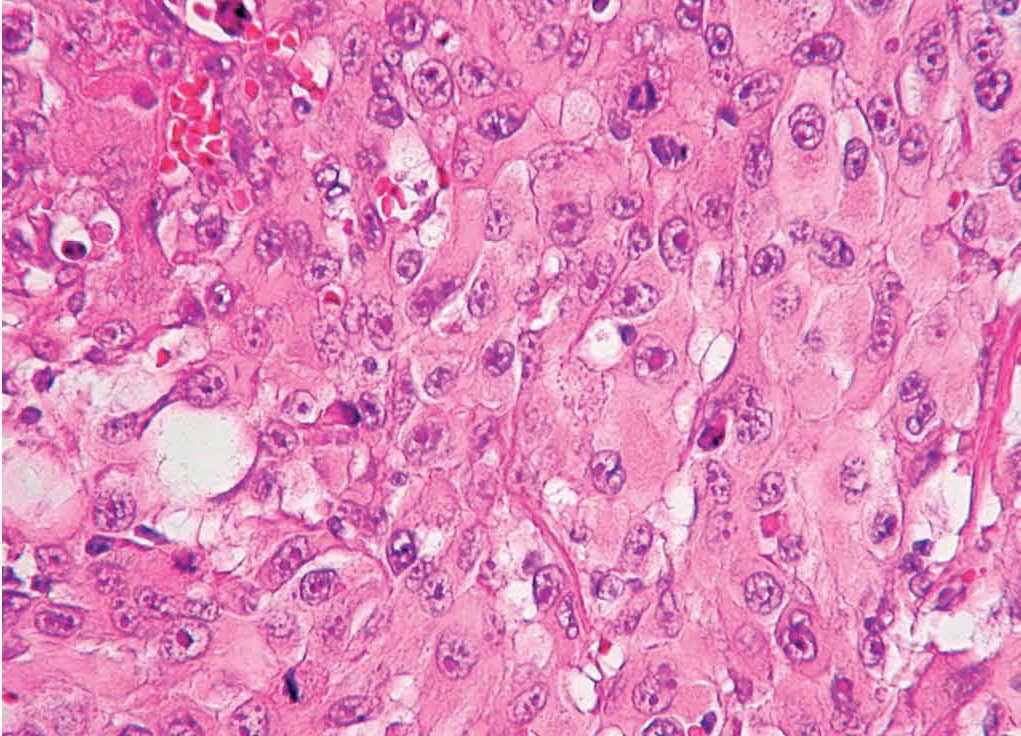
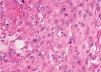
Fig. 6.—En algunos campos predominan las células de aspecto histiocitario, con citoplasma amplio, eosinófilo, núcleo vesiculoso y nucleolo prominente. (Hematoxilina-eosina, ×400.)
En todos los tumores se observaron numerosas mitosis, algunas de ellas atípicas, así como células bizarras y pleomórficas que eran más abundantes cuanto mayor era el índice mitótico.
Se realizó inmunohistoquímica observándose en todos los casos una marcada inmunoexpresión para vimentina, junto con negatividad para citoqueratinas, S100, HMB45, actina, desmina y CD34. El CD68 fue positivo en células de hábito histiocitario.
DISCUSIÓN
Nuestros datos coinciden en general con lo descrito en la literatura médica2,4,5 excepto por una mayor preponderancia masculina. Se ven afectadas básicamente personas de edad avanzada con lesiones localizadas en áreas fotoexpuestas. Sin embargo, hay un paciente de 25 años con una lesión única, de aspecto angiomatoso en tronco, lo cual está descrito como una forma infrecuente de presentación, sobre todo en gente joven. No obstante, algunos autores consideran que estos fibroxantomas atípicos que se desarrollan en miembros y en tronco de gente joven (zonas no fotoexpuestas) y que generalmente no están asociados con daño actínico deberían incluirse en el espectro del denominado histiocitoma fibroso atí-pico6,7 .
El tiempo hasta el diagnóstico es escaso, con una media de 4 meses, lo que coincide con otras series8 . Habitualmente se trata de lesiones únicas, y sólo hay dos publicaciones de localizaciones múltiples9,10 . La morfología de las lesiones es muy variada, pudiendo recordar a diversos tumores, que aparecen en la misma localización con mayor frecuencia, como carcinomas basocelulares o epidermoides. Incluso se ha descrito una variante pigmentada, en la que la pigmentación se debe a la ingesta y posterior degradación de eritrocitos por parte de las células neoplásicas, lo que conduce a la acumulación de hemosiderina en su citoplasma. Esta variante puede ser confundida tanto clínica como histológicamente con el melanoma11 . Por ello es muy complicado realizar el diagnóstico antes del estudio histológico.
El pronóstico en general es bueno, se cree que por la rapidez al diagnóstico y la localización superficial de la tumoración. No obstante, puede haber recidivas, que varían del 5 al 16 % y que suelen deberse a una resección incompleta de la lesión2,12,13 . Se han descrito algunos casos de metástasis, sobre todo ganglionares, pero también a distancia, aunque es una complicación muy rara14,15 . De hecho, Weedon, en su serie de 300 casos no describe ningún caso2 . Sólo uno de nuestros casos presentó recurrencia, la cual fue reextirpada sin recidiva posterior y ninguno ha presentado metástasis. No obstante, son datos a tomar con cautela por el número no muy elevado de casos y el escaso tiempo de evolución. Se han descrito una serie de factores de riesgo de peor pronóstico como el gran tamaño de la lesión, la afectación del tejido celular subcutáneo, marcado pleomorfismo celular, numerosas mitosis atípicas, invasión vascular o perineural, necrosis tumoral y recurrencias locales repetidas3,16 . La histología de la única lesión que recidivó coincidía básicamente con la del tumor primitivo, pero en éste había invasión perineural que, como se ha comentado, es un factor de mal pronóstico para recurrencias y/o metástasis. Es destacable que eran los 3 pacientes de nuestra serie en los que se afectaba el tejido celular subcutáneo en los que se observaba mayor pleomorfismo celular y mayor número de mitosis atípicas, por lo que pueden constituir un grupo de pacientes a vigilar más de cerca.
Todos nuestros pacientes fueron diagnosticados de fibroxantomas atípicos en base a los hallazgos histopatológicos, con presencia de diversos tipos celulares, con predominio del componente fusocelular, dispuestos en un patrón vagamente estoriforme con células bizarras y mitosis atípicas. El diagnóstico de sospecha se confirmó mediante la realización de un panel de inmunohistoquímica, fundamental a la hora de diagnosticar fibroxantomas atípicos, pues este es un diagnóstico de exclusión y es preciso descartar otros tumores fusocelulares que pueden adoptar una morfología similar, con vistas a evitar tratamientos innecesarios que aplicaríamos en tumores más agresivos (tabla 3).
La histogénesis del tumor es incierta, pero parece recaer en una célula mesenquimatosa indiferenciada que podría diferenciarse hacia una línea fibrohistiocitaria o miofibroblástica; de hecho, algunos fibroxantomas atípicos pueden marcar con actina 5,17 . Esto estaría avalado por las tinciones de inmunohistoquímica, ya que aunque posible, sería excepcional que un carcinoma epidermoide dejara de marcar con todas las citoqueratinas. Además en la microscopia electrónica se objetivan células fibroblásticas, histiocitarias y de transición entre ambas 18-20 . La misma célula sería la originaria del histiocitoma fibroso maligno, que es considerado hoy en día como parte del mismo espec-tro 2,3 . El histiocitoma fibroso maligno es el sarcoma de partes blandas más frecuente de la edad adulta, con una supervivencia a los 5 años que varía del 15 al 30 % 21 . El fibroxantoma atípico sería una variedad que se originaría en dermis o en subcutis y que tendría mejor pronóstico.
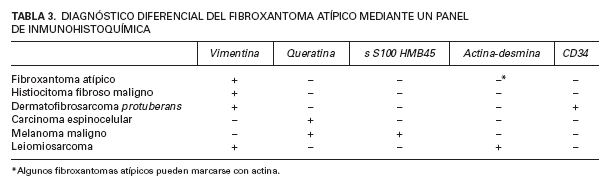
TABLA 3. DIAGNÓSTICO DIFERENCIAL DEL FIBROXANTOMA ATÍPICO MEDIANTE UN PANEL DE INMUNOHISTOQUÍMICA
En ocasiones puede ser difícil diferenciar entre un fibroxantoma atípico o un histiocitoma fibroso maligno superficial, pues muchas veces es una cuestión de grado. De hecho, para algunos autores, cualquier neoplasia que afecte extensamente el subcutis, fascia o músculo, o que presente extensa invasión vascular o necrosis correspondería a un histiocitoma fibroso maligno 3 . Otros datos que deben hacer sospechar un histiocitoma fibroso maligno es que haya un patrón claramente estoriforme o un número muy elevado de mitosis atípicas. La tinción con CD74/ LN-2 es positiva en el 90 % de los histiocitomas fibrosos malignos frente al 10 % de los fibroxantomas atípicos y puede ayudar a distinguirlos, aunque su significado patogénico y diagnóstico todavía no está del todo claro 22 .
La etiología de los fibroxantomas atípicos es desconocida, pero el hecho de que la mayoría de ellos ocurra en personas de edad avanzada y sobre una piel con daño actínico hace pensar en la radiación ultravioleta como un factor causal debido al daño irreparable del ADN de células mesenquimatosas. Hay varios trabajos que inciden en este aspecto, destacando la importancia de la alteración de la p5323,24 . De hecho en 9 de nuestros 10 pacientes se encontraban queratosis actínicas acompañantes y en el 50 % al menos otro carcinoma cutáneo, destacando 2 pacientes con 5 y 6 carcinomas basocelulares, respectivamente, lo que habla a favor del factor patogénico de la radiación ultravioleta. También se ha implicado la radioterapia previa, al igual que en otros sarcomas, como ocurrió en uno de nuestros pacientes que había recibido radioterapia por un carcinoma epidermoide de mejilla 15 años antes.
El tratamiento del fibroxantoma atípico es quirúrgico, con un muy buen pronóstico si se elimina la lesión en su totalidad. No está claro el papel de la cirugía de Mohs en estos casos, debido a la escasez de las series, las cuales proporcionan a su vez resultados muy diversos12,13,25,26 . En teoría es un tumor que debería beneficiarse de esta técnica, pero la escasez de estudios hace que no pueda recomendarse en el momento actual como tratamiento de primera elección.
Otro problema es que se trata de lesiones habitualmente de pequeño tamaño, que no se diagnostican inicialmente de fibroxantoma atípico con lo que son extirpadas y el diagnóstico se realiza a posteriori , siendo muy complicado utilizar la cirugía de Mohs. En resumen, hemos presentado una serie de pacientes con fibroxantona atípico, cuyos datos clínicos, epidemiológicos y evolutivos coinciden en general con lo descrito en la literatura médica. A pesar de ser un tumor infrecuente debe ser tenido en mente y realizar un adecuado diagnóstico mediante técnicas de inmunohistoquímica para evitar tratamientos agresivos e innecesarios.
Correspondencia:
Enrique Gómez de la Fuente. Servicio de Dermatología. Fundación Hospital Alcorcón. Budapest, 1. 28922 Alcorcón. Madrid. España. egomezf@fhalcorcon.es
Recibido el 29 de julio de 2004. Aceptado el 15 de diciembre de 2004.


