



INTRODUCCION
La facomatosis pigmentovascular (FPV) fue descrita por Ota en 1947 como la asociación de un nevo vascular telangiectásico con nevo pigmentario y verrugoso1, y desde entonces se han descrito más de 120 casos. En 1979, Hasegawa y Yasuhara proponen la primera clasificación, dividiéndola en cuatro tipos. En 1987, Ruiz-Maldonado describió 4 pacientes con alteración vascular extensa, pigmentación oculocutánea y alteración neurológica grave, y propuso el término de «facomatosis pigmentovascular» para referirse a un nuevo síndrome neurocutáneo2. La FPV es la asociación de una anomalía pigmentaria extensa junto a una vascular también extensa, que pueden estar acompañadas de otras lesiones cutáneas o sistémicas. Se establecen cuatro tipos (tabla 1), de los que el tipo II es el más frecuente (> 85 %)3-6. Algunos autores han criticado esta clasificación, señalando que todos los tipos y subtipos se explicarían por una misma teoría genética. La mayor parte de los pacientes son de origen oriental o hispano.

Además de las anomalías cutánea que provocan el síndrome, algunos pacientes presentan anomalías extracutáneas asociadas (tabla 2). Las más habitualmente descritas son las asociadas al síndrome de Sturge-Weber7, como angiomatosis leptomeníngea, calcificaciones intracraneales, atrofia cerebral, retraso mental, convulsiones, glaucoma y buftalmos8. Otras menos frecuentes son síndrome de Klippel-Trenaunay, enfermedad de Moya-Moya e hipoplasia de sistema porta y varices esofágicas. La pigmentación aberrante puede asociarse a nevos de Ota, melanosis de las escleróticas y nevos de Ito y ocasionalmente se han descrito asimetría de las córneas y pigmentación retiniana5,9. Otras manifestaciones asociadas esporádicamente son deficiencia de immunoglobulina A (IgA)10, agenesia de riñón11, hemangioma renal12 y atrofia e hipertrofia de tejido subyacente13.

DESCRIPCION DEL CASO
Una lactante de 5 meses de edad, sin antecedentes familiares ni personales de interés, fue remitida a nuestra consulta para valoración de unas manchas cutáneas que presentaba desde el nacimiento. El embarazo, parto y período perinatal habían transcurrido sin incidencias, y la niña presentaba un desarrollo psicomotor adecuado a su edad. En la exploración física se observó una mancha eritematosa telangiectásica extensa que afectaba a hemicuerpo izquierdo desde la cabeza a la cintura, hombro derecho, tronco y genitales externos (fig. 1). Las lesiones se detenían en la línea media a nivel facial, incluyendo mucosa oral, pero no respetaban la línea media en el tronco y región vulvar. En el brazo izquierdo, la mancha eritematosa se detenía bruscamente en la cara anterolateral. Junto a esta mancha, se apreció una extensa pigmentación de color azul grisácea que afectaba al abdomen, glúteo izquierdo y raíz de muslo izquierdo, en forma de grandes manchas de morfología poligonal, que remedaban un patrón en tablero de ajedrez. No se apreciaron alteraciones en la pigmentación del globo ocular.
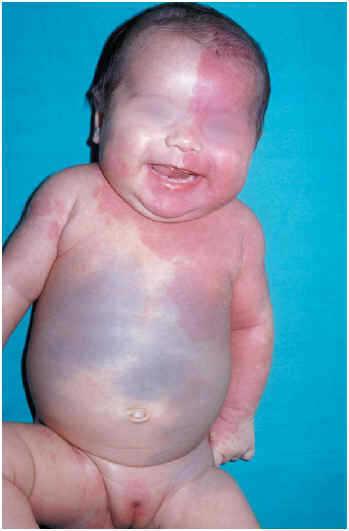
Fig. 1.--Mancha eritematosa telangiectásica que afecta a hemicuerpo izquierdo.
Se practicaron exploración física general, exploración neurológica y oftalmológica incluyendo fondo de ojo, ecografía transfontanelar, resonancia magnética craneal y electroencefalograma. Todas estas pruebas ofrecieron resultados compatibles con la normalidad. En el seguimiento de la paciente a los 6 meses no se observaron cambios en las lesiones descritas.
DISCUSION
Nuestra paciente presentaba un cuadro clínico compatible con una FPV tipo IIa. La patogenia de la asociación de las extensas anomalías vasculares y pigmentarias en un mismo paciente es desconocida, aunque se ha propuesto para su explicación un mecanismo conocido como manchas gemelas14. Las manchas gemelas son manchas de tejido mutante que difieren entre sí y del fondo de tejido sano circundante. Se supone que, partiendo de una célula heterozigota para dos mutaciones distintas entre sí, situadas en el mismo cromosoma, un fenómeno de recombinación somática entre cromosomas homólogos producida durante la mitosis puede dar lugar a dos células hijas que son homozigotas para las dos mutaciones iniciales. Las manchas gemelas pueden deberse a mutaciones que suceden en el mismo alelo o en distintos alelos, pero obligatoriamente próximos entre sí o al menos en el mismo cromosoma, como ya hemos señalado15. Las manchas gemelas alélicas, desde el punto de vista clínico, se manifiestan como áreas de «exceso y defecto» dependiendo de la cualidad que determine el gen mutado, como por ejemplo la asociación de nevo anémico con nevo telangiectásico o la asociación de un área de hipopigmentación yuxtapuesta a un área de hiperpigmentación o cutis tricolor14-16. Las manchas no alélicas se deben a mutaciones en alelos diferentes y manifiestan dos tipos de lesiones distintas que aparecen en el mismo individuo. Sin embargo, la gran variedad y cantidad de asociaciones diferentes de lesiones descritas como manchas gemelas no alélicas puede ir en contra de esta teoría, ya que ésta implicaría que todas las mutaciones posibles se encuentran muy próximas entre sí en el mismo cromosoma, lo cual es altamente improbable. Las manchas gemelas pueden manifestarse con un patrón en mosaico de cualquiera de sus tipos, aunque éste no es obligatorio. Sea como fuere, el patrón de distribución de estos cuadros, la asociación de tejido cutáneo mutante y la posible afectación extracutánea hacen suponer que este tipo de anomalías del desarrollo de la piel no sea meramente casual. Por otra parte, es frecuente la asociación con anomalías extracutáneas. Concretamente, en la FPV deben realizarse exploraciones neurológicas y oftalmológicas detalladas, incluyendo una resonancia magnética craneal y un electroencefalograma.


