





Ochronosis is a disease in which ochre pigment is deposited in the tissues. Two forms have been described: endogenous and exogenous. The 2 forms have different clinical presentations and laboratory alterations, but the histology features are identical.
The endogenous form, also known as alcaptonuria, is of autosomal recessive inheritance and is due to a congenital deficiency of the enzyme homogentisic acid oxidase (HGAO), which is involved in the metabolism of the amino acids phenylalanine and tyrosine. The deficit of HGAO leads to tissue accumulation of homogentisic acid (HGA), which is excreted in high concentrations in the urine and in secretions.1,2
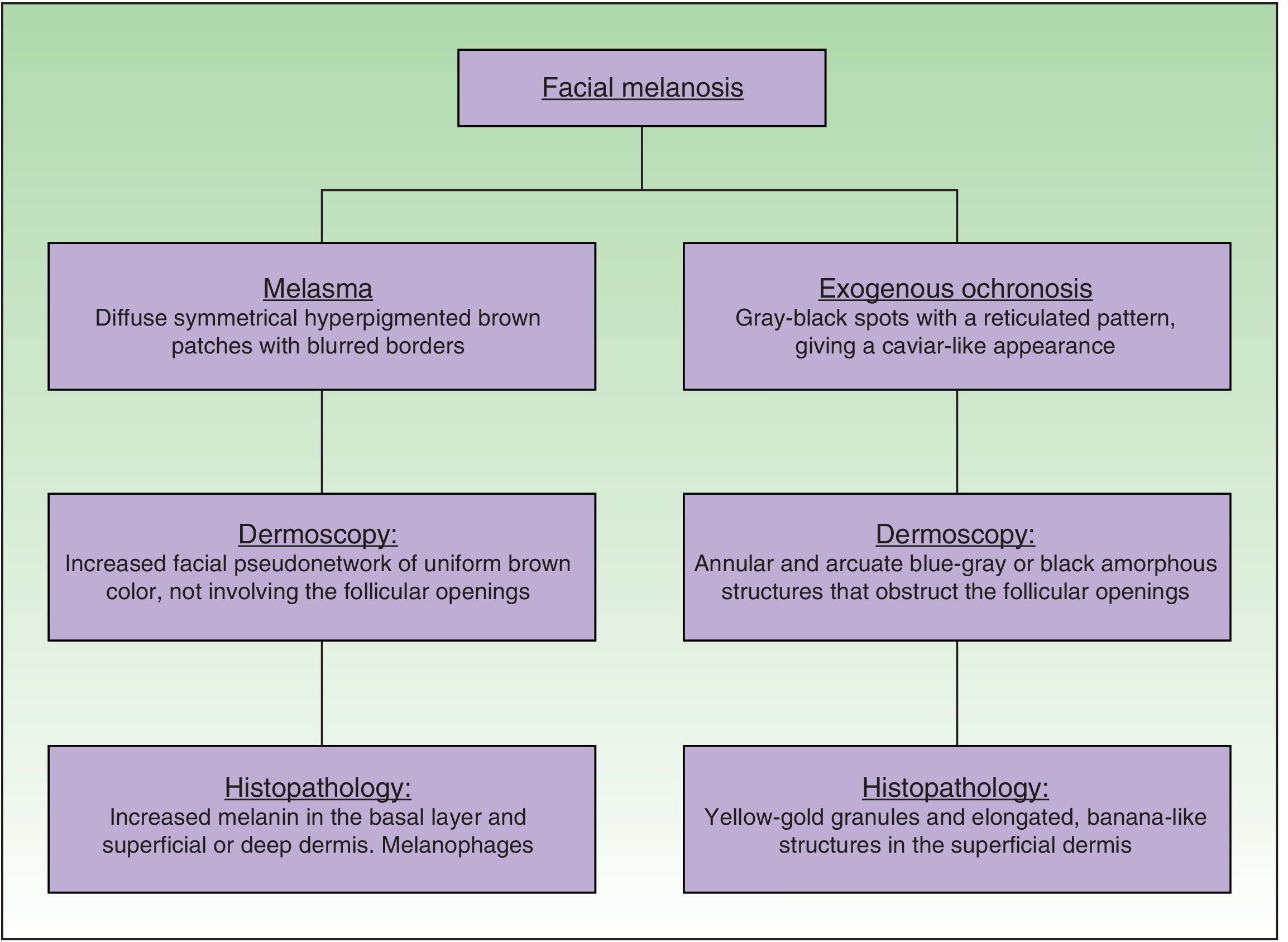
In exogenous ochronosis (EO), deposits of polymerized HGA build up in the superficial dermis due to local inhibition of the enzyme HGAO caused by a prolonged use of topical treatments such as hydroquinone and its derivatives.3
We describe the case of a 49-year-old housewife born and resident in Leon, Guanajuato, Mexico. In her background we detected a 2-year history of hypothyroidism. She consulted for a 16-year history of hyperchromic macules on both her cheeks and on her forehead. For the previous 15 years, the macules had been treated with hydroquinone for long but intermittent periods. She had also used home remedies and sunscreen without observing any improvement.
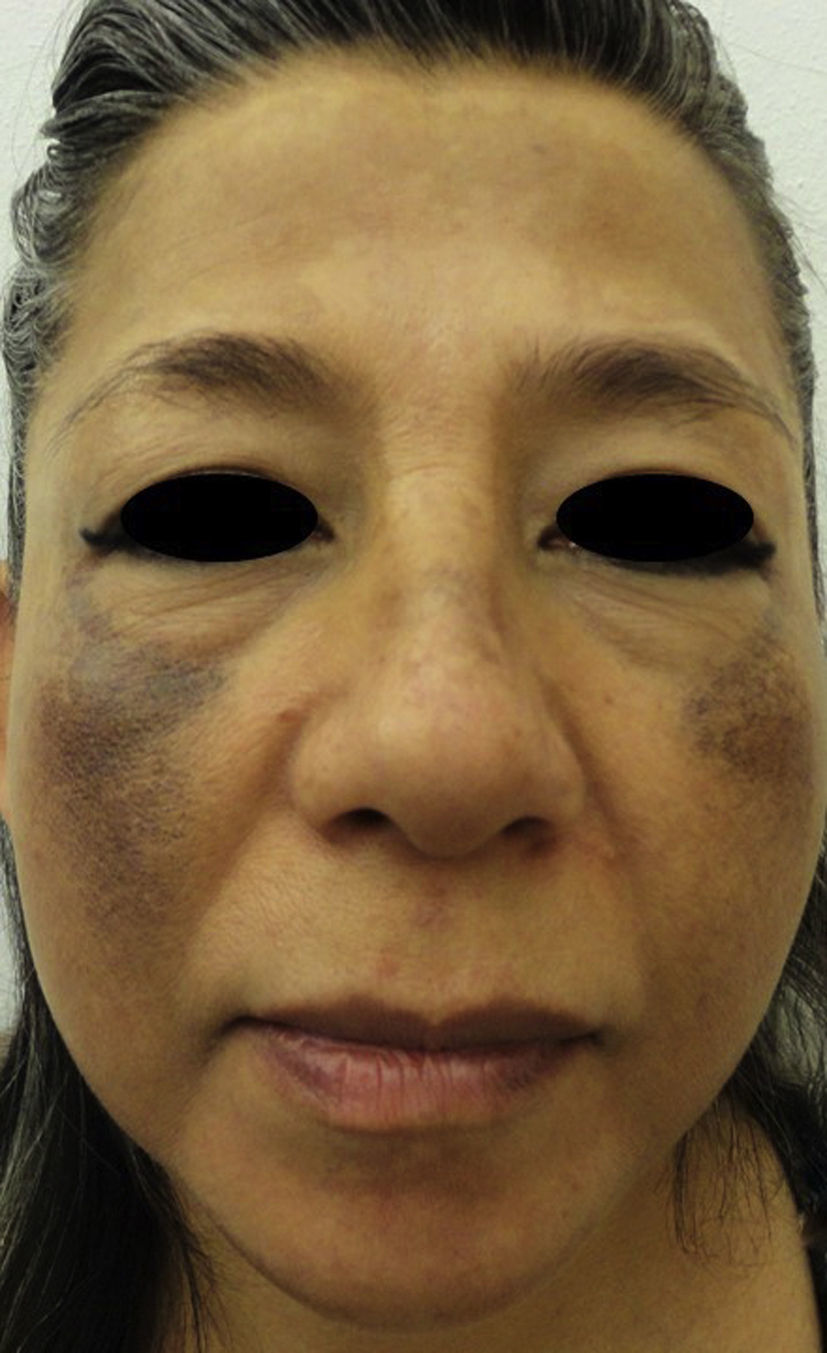
On examination, a symmetrical, bilateral facial dermatosis was observed affecting both cheeks and the dorsum of the nose, but respecting the periocular region, the nasolabial fold, and the perioral area. The dermatosis consisted of diffuse dark brown macules on the forehead and cheeks, and hyperpigmented dark gray spots. The patient denied any symptoms (Figs. 1 and 2A).
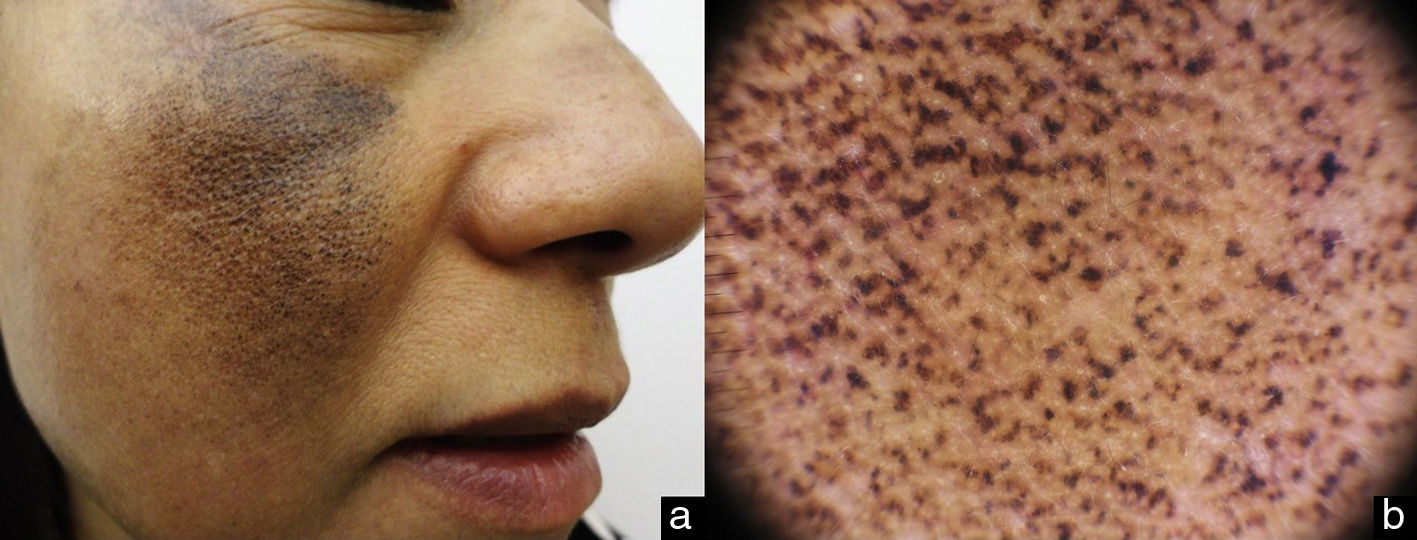
Dermoscopy revealed an increase in the facial pseudonetwork, with intensely pigmented amorphous dark-brown structures with a reticular pattern; these structures obstructed the follicular openings (Fig. 2B).
Histopathology with hematoxylin and eosin stain showed long, banana-shaped deposits of an acellular material of a pale gold color, with a mild lymphohistiocytic interstitial inflammatory infiltrate (Fig. 3, A and B). Fontana-Masson stain highlighted the melanic pigment (Fig. 3C). These findings confirmed the clinical and dermoscopic diagnosis of EO.
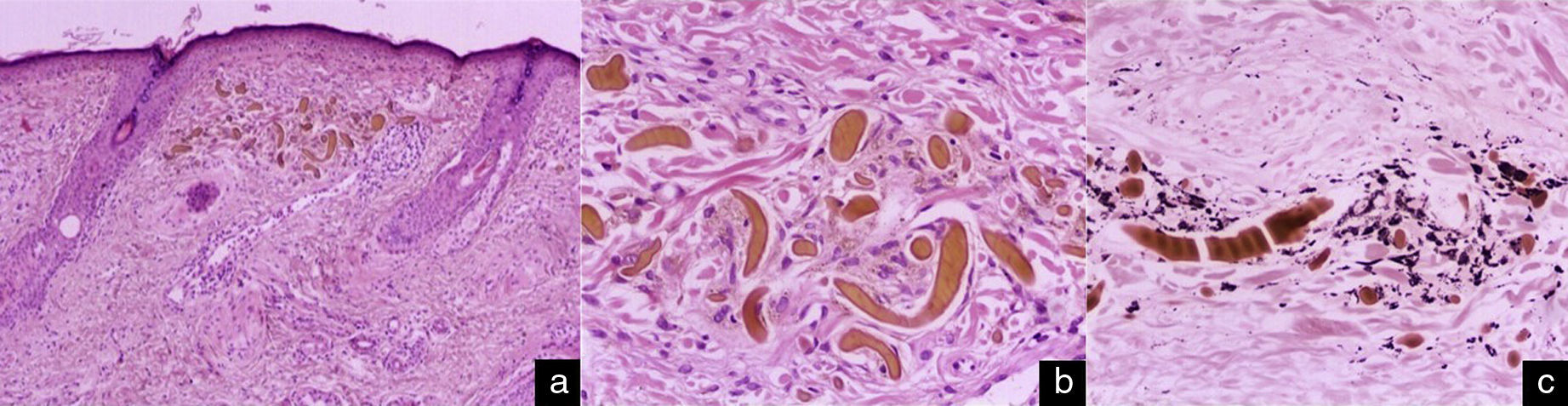
Histology. Elongated banana-shaped deposits of a pale-gold–colored acellular material in the superficial dermis, a typical image of exogenous ochronosis. Hematoxylin and eosin, original magnification A, ×10 and B, ×40. C, Highlighting of the melanic pigment. Fontana-Masson stain, original magnification ×40.
EO presents as a symmetrical bilateral dermatosis that typically affects sun-exposed areas and the skin over bony prominences, most commonly in the malar and temporal regions and area of the mandible and chin. It is characterized by hyperpigmented reticulated macules, of dark gray or grayish brown color, with lighter and darker areas, giving a clinical appearance similar to caviar.3–5
The worldwide prevalence is considered to be low. It is most common in women in the third and fourth decades of life, and particularly affects phototypes III and iv.4,5
The onset of EO has been associated with hydroquinone and also with numerous other substances: topical mercurials, oral and parenteral antimalarial drugs, the application of phenol, resorcinol, or picric acid, and levodopa. However, no single etiologic factor that triggers the disease has been identified. Our patient had been diagnosed 16 years earlier with melasma affecting her frontal and malar regions. However, despite the clear clinical evidence of EO, the patient continued to be prescribed hydroquinone for the treatment of her hyperpigmentation, using it for a total of 15 years.3,6,7
Dermoscopy is a non-invasive method that can aid diagnosis of EO very specifically. The dermoscopic features are the presence of amorphous annular and arcuate structures that are dark blue or grayish-black in color depending on the depth of the pigment in the skin. These structures surround and occasionally obliterate follicles orifices, and there is an accentuation of the normal pseudonetwork of the skin of the face. Our patient presented the characteristic changes, with amorphous and reticulated hyperpigmented structures (Fig. 4).7–9
The differential diagnosis includes melasma, bilateral Ota nevus, drug-induced hyperpigmentation, postinflammatory pigmentation, and dermatosis papulosa nigra.8,10
The definitive diagnosis is made by the detection of deposits of HGA in the superficial dermis in the form of long, banana-like, curvilinear structures of different sizes and of yellow-gold color. Other changes have been reported, including edema and degeneration of collagen fibers and a histiocytic and plasma cell inflammatory infiltrate. Solar elastosis and pigment incontinence are often observed.8,9
It is important for the dermatologist to recognize the clinical presentation and the dermoscopy and histopathology findings of this dyschromia induced by hydroquinone, one of the most common depigmenting agents used in medical practice to treat melasma, and which is available over the counter in many countries. Furthermore, exogenous ochronosis can be confused with other pigment disorders, including melasma itself, and this must be taken into account in the differential diagnosis.
Please cite this article as: Córdova ME, Pérez-Rojas DO, López-Marquet AD, Arenas R. Ocronosis exógena en melasma facial. Actas Dermosifiliogr. 2017;108:381–383.


