





INTRODUCCION
La enfermedad de Whipple (EW) es un proceso poco frecuente, descrito por primera vez en 1907 por George H. Whipple, quien lo denominó lipodistrofia intestinal. Es más frecuente en varones y tiene un pico de incidencia entre los 40 y los 50 años. El microorganismo responsable de la EW es un actinomiceto grampositivo, Tropheryma Whippelii, que ha podido ser cultivado recientemente 1. Se trata de un trastorno sistémico que suele caracterizarse por diarrea, pérdida de peso, malabsorción, fiebre, oligoartritis o poliartritis, linfadenopatías y en ocasiones manifestaciones neurológicas. La afectación cutánea es rara, siendo la más frecuente la hiperpigmentación. La EW sin tratamiento tiene una mortalidad del 100 %, por lo que es necesario realizar un diagnóstico y tratamiento lo más precoz posible 2.
DESCRIPCION DEL CASO
Varón de 48 años, sin antecedentes personales ni familiares de interés, que presentaba cuadro de diarrea crónica de más de un año de evolución y síndrome general, con astenia, anorexia y adelgazamiento de más de 15 kg de peso que le confería un aspecto caquético, con poliadenopatías de 2-3 cm de diámetro, elásticas y no adheridas a planos profundos en cuello, axilas y región inguinal. Posteriormente, desarrolló un cuadro de demencia subcortical, con cambios en el comportamiento, déficit de memoria, ataxia y hemiparesia IV/V derecha. En la exploración cutánea se observaron encías edematosas que sangraban con facilidad (fig. 1), lesiones purpúricas en piernas, de predominio folicular (fig. 2), así como descamación cutánea generalizada ictiosiforme (fig. 3). En las pruebas complementarias destacaban hemoglobina 9,2 g/dl; volumen corpuscular medio 100 fl; 2.500 leucocitos con fórmula normal; velocidad de sedimentación globular 55 mm y datos de malnutrición severa con prealbúmina 8 mg/dl (normal 10-40) y retinol 1,3 mg/dl (normal 3-6); vitamina B12 171 pcg/ml (normal 211-911), ácido fólico 0,5 ng/ml (normal > 1,6); niveles séricos de ácido ascórbico 7 micromol/l (normal 20-80 micromol/l); enzima conversora de angiotensina 71,3 U/l (normal 8-55); proteinograma con Ig M < 30 mg/dl; serologías hepatitis B, C, lúes y virus de la inmunodeficiencia humana, negativas; coprocultivos y parásitos en heces, negativos; bacilos ácido-alcohol resistentes en esputo y orina, negativos; Mantoux 0 mm; bioquímica y recuento celular de líquido cefalorraquídeo normal; radiografía de tórax normal; resonancia magnética nuclear cerebral con áreas extensas de desmielinización de sustancia blanca supra e infratentorial. Se realizó endoscopia del tubo digestivo con estudio anatomopatológico, observando a nivel del estómago gastritis crónica activa y la presencia de granulomas epitelioides no necrotizantes a nivel del duodeno, colon e íleon, sin observarse bacilos ácido-alcohol resistentes. La biopsia de un ganglio linfático axilar demostró una linfoadenitis granulomatosa no necrotizante. El estudio histológico de las lesiones purpúricas de las piernas mostró folículos pilosos con hiperqueratosis folicular y discreta hemorragia perifolicular (fig. 4). En la biopsia de lesiones de piel de tórax se observó una epidermis ligeramente adelgazada con hiperqueratosis de apariencia laminar sin paraqueratosis y atenuación de la capa granulosa (fig. 5). Con todos estos datos inicialmente se estableció el diagnóstico de enfermedad granulomatosa idiopática generalizada (probable sarcoidosis) y se instauró tratamiento corticoideo oral, con escasa mejoría del cuadro clínico. Posteriormente, ante la escasa respuesta al tratamiento, se revisaron todas las biopsias, y aunque no se encontraron macrófagos PAS positivos en las mismas, se enviaron muestras de biopsia duodenal, sangre y líquido cefalorraquídeo para la determinación de fragmentos genómicos de Tropheryma whippelii mediante reacción en cadena de la polimerasa (PCR) y para microscopía electrónica, que fueron positivos en las muestras duodenales. Ante estos resultados se estableció el diagnóstico de enfermedad de Whipple, forma granulomatosa pseudosarcoidótica, con malabsorción intestinal secundaria que condicionó cuadro de malnutrición extrema, escorbuto, ictiosis adquirida y anemia megaloblástica. Se instauró tratamiento con imipenem más estreptomicina durante 15 días, seguido de trimetropim-sulfametoxazol, así como complejos vitamínicos con vitamina C y vitamina B12. A las pocas semanas se produjo una completa resolución de las lesiones cutáneas y de la diarrea, así como ganancia de 10 kg de peso. En el control gastroscópico al mes del tratamiento no se observaron lesiones macro ni microscópicas. A pesar de completar el tratamiento durante dos años el paciente sólo presentó discreta mejoría desde el punto de vista neurológico.
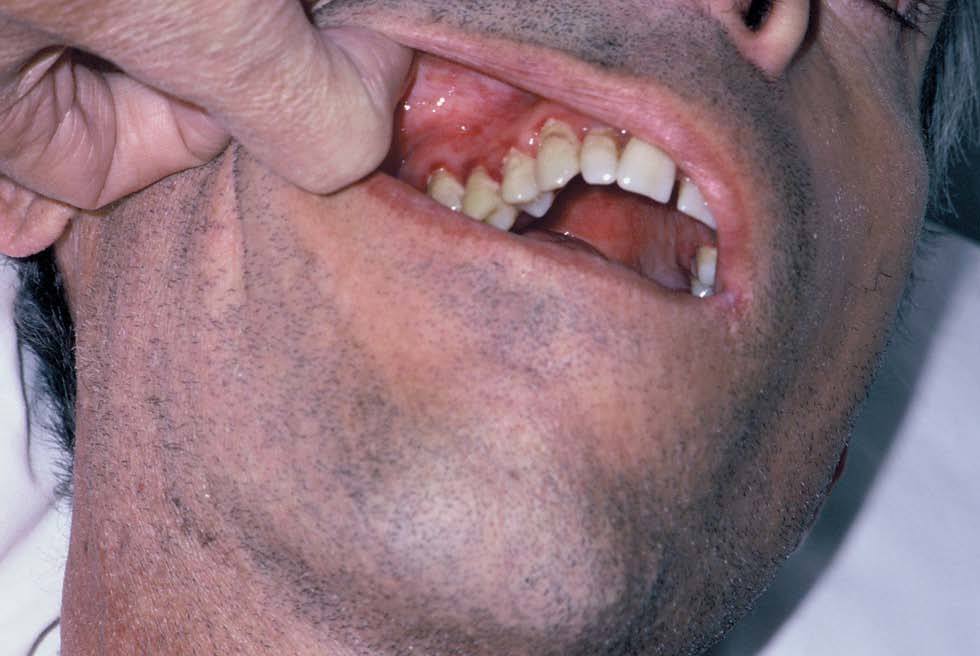
Fig. 1.--Hemorragias gingivales.
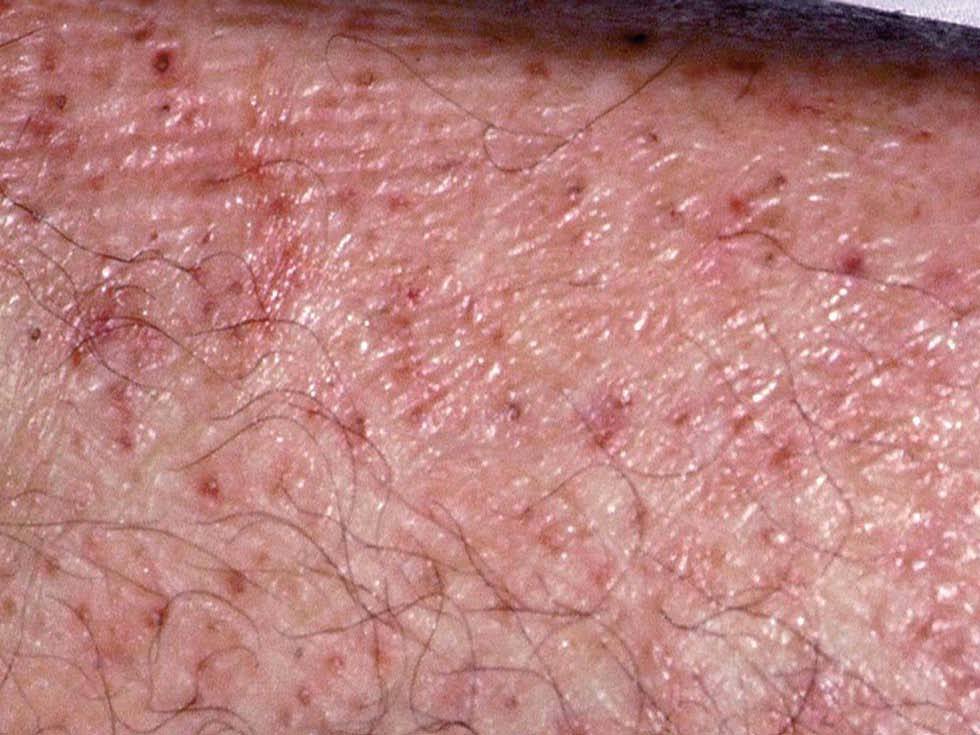
Fig. 2.--Lesiones purpúricas hiperqueratósicas de predominio
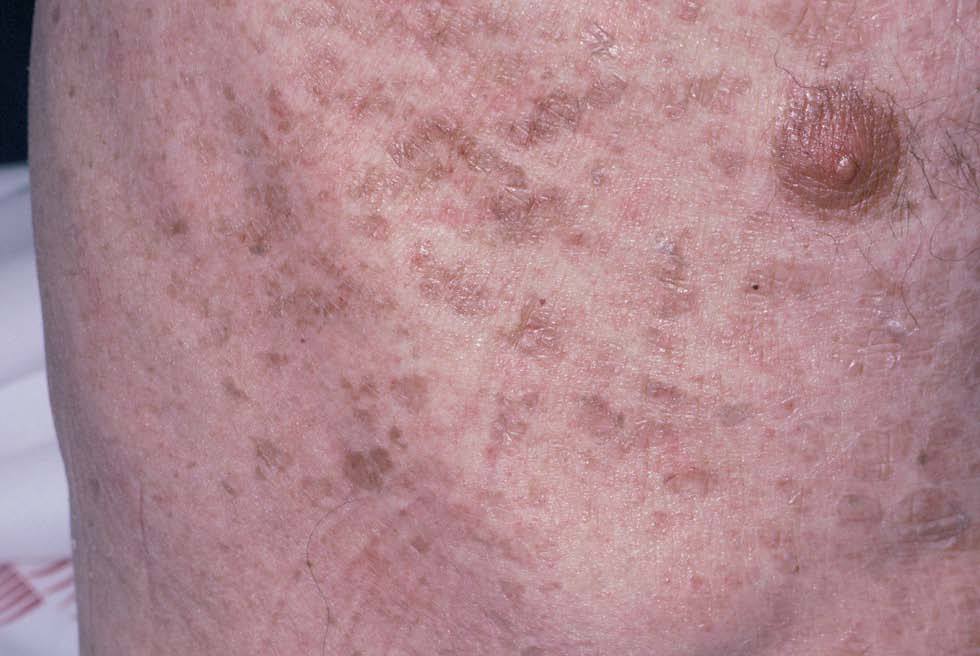
Fig. 3.--Ictiosis adquirida.
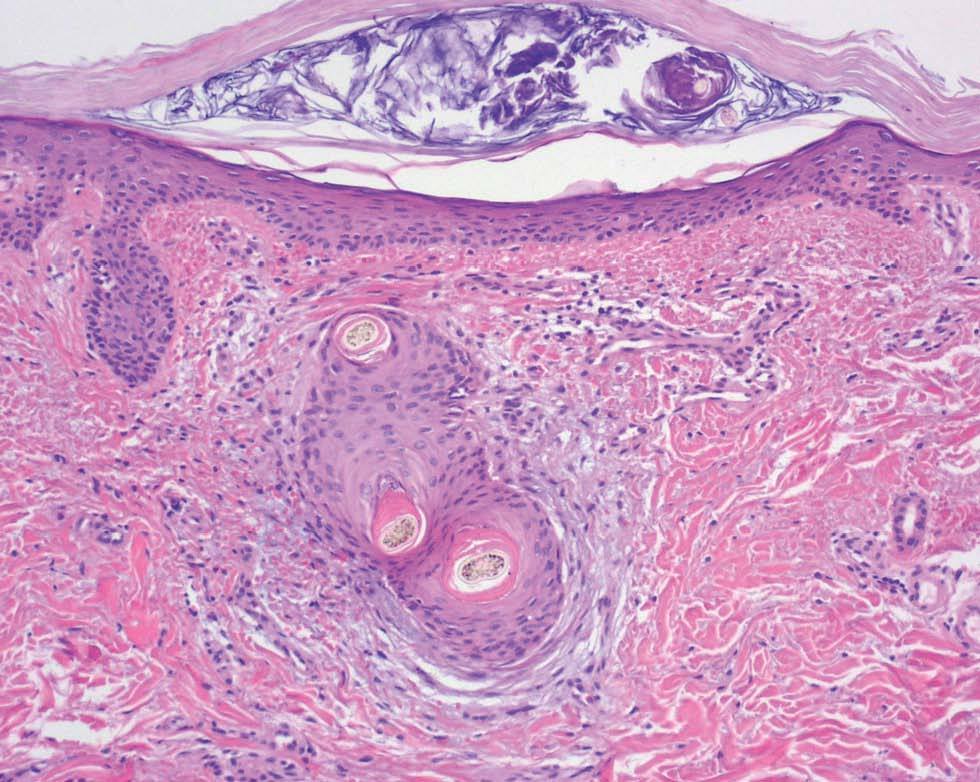
Fig. 4.--Pelos retorcidos, hiperqueratosis folicular y extravasación hemática (hematoxilina-eosina, 200).
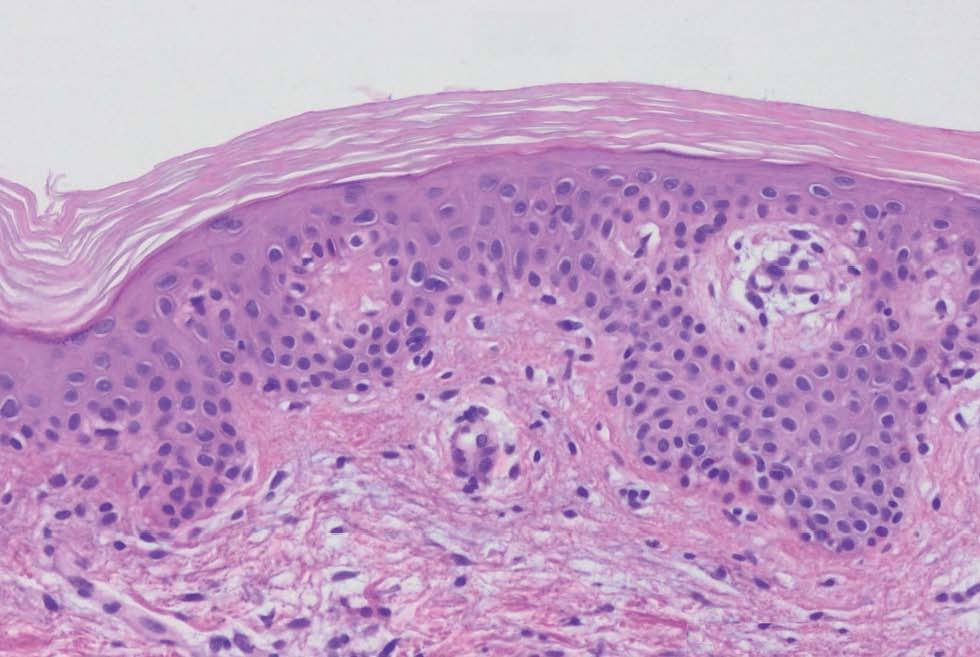
Fig. 5.--Hiperqueratosis y atenuación de la capa granulosa (hematoxilina-eosina, 400).
DISCUSION
La EW es una enfermedad sistémica rara, de comienzo insidioso y difícil de diagnosticar, que tiene predilección por el sistema digestivo, especialmente el intestino delgado. Probablemente se trate de un proceso infradiagnosticado, aunque el número de casos ha aumentado desde que podemos identificar el agente responsable mediante PCR 2. El microorganismo causal (Tropheryma whippelii) se caracterizó molecularmente en 1991 mediante PCR y en el año 2000 se logró cultivar en células HLE (línea celular de fibroblastos humanos) 1. Su modo de transmisión no ha sido identificado. Es posible que en la patogenia intervenga cierto déficit inmunitario y susceptibilidad individual del huésped. El espectro de síntomas clínicos de esta enfermedad es muy variado, y en ocasiones inespecífico, lo que puede dificultar su diagnóstico. En lo que a éste se refiere, la endoscopia oral con biopsia duodenal problablemente sea la exploración más útil y rentable, debido a que la afectación intestinal comienza en la primera porción duodenal. Actualmente las técnicas moleculares que utilizan los métodos de amplificación genética mediante PCR permiten identificar el bacilo en los tejidos, en fluidos orgánicos o en células mononucleares de sangre periférica 3. La histología típica consiste en la afectación de la lámina propia por histiocitos espumosos con abundante material granular PAS positivo y resistente a la diastasa, que corresponde a bacterias parcialmente degradadas. Estos hallazgos también podemos verlos a nivel de corazón, pulmón, bazo, hígado, médula ósea, glándulas suprarrenales y riñones, entre otros 4. Sin embargo, no es infrecuente que los pacientes desarrollen una respuesta granulomatosa tipo sarcoideo con tinción PAS negativa, lo que puede llevar a un diagnóstico de sarcoidosis 5-8. Las formas pseudosarcoidóticas de la enfermedad fueron ya descritas por Newman y Pope en 1948 9. En 1987 Wilcox et al comprobaron que los granulomas PAS negativos eran debidos a EW, pues el estudio con microscopía electrónica reveló bacilos en los espacios intercelulares e intracelulares, lo que fue confirmado posteriormente por Spapen et al 10,11.
Los hallazgos cutáneos de la EW, a excepción de la hiperpigmentación, son poco frecuentes y habitualmente inespecíficos 12. Se han descrito nódulos subcutáneos, eritema nodoso, eritrodermia, púrpura, vasculitis, hiperqueratosis, lesiones urticariformes, erupciones liquenoides y dermatitis eczematosas 6,13. La afectación cutánea específica en la EW es extraordinariamente rara. En la literatura revisada hemos encontrado pocos casos descritos. Todos ellos presentaban lesiones nodulares subcutáneas, solitarias o múltiples con histología de paniculitis septal, con un gran número de histiocitos espumosos con abundante material granular PAS positivo y resistente a la diastasa, superponibles a los hallazgos encontrados en biopsias del tubo digestivo de EW 2,4,14-16.
En los síndromes de malabsorción, un 20 % de los pacientes presentan manifestaciones dermatológicas, directamente causadas por el déficit en la absorción de determinados nutrientes 13. Nuestro paciente desarrolló lesiones cutáneas de escorbuto e ictiosis adquirida. El déficit de vitamina C produce el escorbuto, que clínicamente se caracteriza por hiperqueratosis folicular, pelos en sacacorchos y hemorragias perifoliculares, que son más evidentes en antebrazos, abdomen y piernas, junto con encías edematosas, friables y sangrantes. Además, las heridas cicatrizan con dificultad y también podemos encontrar petequias, equimosis y hemorragias subungueales. El desarrollo de ictiosis en la vida adulta puede ser, asimismo, una manifestación de malabsorción; pero también se ha descrito en asociación con neoplasias malignas, fármacos, enfermedades endocrinológicas y metabólicas, sarcoidosis, infección por el virus de la inmunodeficiencia humana (VIH) y enfermedades autoinmunes 13.
Inicialmente el tratamiento de la EW requiere terapia antibiótica parenteral durante 2 a 4 semanas con distintos antibióticos como penicilina, estreptomicina o cefalosporinas de tercera generación, como la ceftriaxona. Posteriormente se instaura tratamiento antibiótico con cotrimoxazol o tetraciclinas durante períodos superiores a un año para evitar recidivas. Con el tratamiento los síntomas mejoran rápidamente, pero la recuperación completa de los tejidos puede requerir más de dos años 2.
Consideramos que ante un enfermo diagnosticado de enfermedad granulomatosa idiopática con cuadro de patología gastrointestinal y de malabsorción, con mala respuesta al tratamiento, debemos incluir en el diagnóstico diferencial la EW y realizar la determinación de secuencias específicas de Tropheryma Whippelii mediante PCR, aunque en las biopsias no encontremos macrófagos PAS positivos. La realización de microscopía electrónica también es de utilidad en el diagnóstico de EW, para demostrar la presencia de bacilos en los granulomas.
Exponemos el caso de un paciente con enfermedad de Whipple de forma pseudosarcoidótica que desarrolló como manifestaciones cutáneas, secundarias al síndrome de malabsorción, escorbuto e ictiosis adquirida. Sólo hemos encontrado en la literatura un caso de escorbuto asociado a EW 17 y ninguno de ictiosis adquirida asociado a esta enfermedad.
Correspondencia:
Yolanda Juárez Casado.
San Roque, 25, 3.º D.
27002 Lugo. España.
yojuarezca@yahoo.es
Recibido el 28 febrero de 2006.
Aceptado el 27 de junio de 2006.


