







INTRODUCCIÓN
La esclerosis tuberosa es una entidad clínica que se clasifica dentro del grupo de las facomatosis o síndromes neurocutáneos, en el cual se incluyen neurofibromatosis tipos I y II, síndrome de Von Hippel-Lindau y enfermedad de Sturge Weber. Se caracteriza por la aparición de múltiples hamartomas y se distingue de ellos por numerosas características diferenciales, siendo la más significativa la afectación de casi todos los órganos y sistemas. El mecanismo fisiopatológico responsable no se conoce en última instancia, pero algunas formas escapan al desarrollo polivisceral y determinan las formas fustres o incompletas. Afecta por igual a todos los sexos y razas, aunque algunas series señalan un ligero predominio en varones y su incidencia poco frecuente en la raza negra.
Las primeras referencias de la enfermedad corresponden a Virchow1 quien en 1860 describió escleromas en el cerebro, pero fue Vogt2 quien definiría la tríada clásica «adenomas sebáceos», retraso mental y epilepsia». Los hallazgos oculares de la enfermedad, a los que llamaría facomas se deben a Van der Hoeve3 y así introdujo el término facomatosis en 1921. En 1942 las diferentes manifestaciones clínicas de la enfermedad quedaron finalmente englobadas al constituir el «complejo esclerosis tuberosa» gracias a Moolten4.
La esclerosis tuberosa se debe a un trastorno disembrioplásico que afecta a las tres hojas blastodérmicas con un patrón de herencia autosómico dominante de expresividad variable y penetrancia incompleta que se traduce clínicamente por la aparición de múltiples hamartomas. La prevalencia de la esclerosis tuberosa se estima entre 1/5.800 y 1/15.000, lo cual hace de ella una de las enfermedades genéticas más frecuentes. Su fisiopatología no está bien determinada, aunque a grandes rasgos el trastorno radica en la migración, proliferación y diferenciación celular5, habiéndose identificado células anormales que asemejan parcialmente neuronas y parcialmente astrocitos (N-cells) tanto en los túberes cerebrales como en las lesiones cutáneas procedentes de la cresta neural durante la embriogénesis6.
BASES GENÉTICAS Y MOLECULARES
Este proceso es muy variable en cuanto a sus manifestaciones clínicas, y su expresividad parece estar condicionada por las mutaciones experimentadas. La frecuencia de mutaciones parece oscilar entre el 50% y el 70%, lo que, como veremos, hace el consejo genético poco útil. Es importante explorar a los padres para un adecuado consejo genético, ya que si puede demostrarse que son portadores sería un dato a tener en cuenta de cara a futuros embarazos. Es muy raro que aquellas personas que padecen una forma oligosintomática no tengan algún signo cutáneo característico. En caso de no hallarse ninguno es recomendable realizar un fondo de ojo (con dilatación), una tomografía axial computarizada (TAC) craneal y una ecografía renal. La resonancia magnética nuclear (RMN) puede dar falsos positivos y además no puede evidenciar la presencia de calcio7. El objeto de las mutaciones radica en dos loci diferentes8: 9q34 (que codifica la proteína denominada hamartina) y 16p13.3 (que codifica la proteína denominada tuberina), y por ello hablamos de esclerosis tuberosa tipo 1 y 2, respectivamente9. Los estudios de ligamiento se iniciaron de manera precoz en la década de los ochenta y fueron difíciles debido a la ausencia de largas series familiares. En 1987 se demostró el ligamiento de los genes de la esclerosis tuberosa con el grupo ABO en el cromosoma 9p34, ya mencionado; sin embargo, no estaban presentes en todas las familias. Tambien se había descrito ligamiento en otros cromosomas (11q y 12q), aunque otros grupos de investigación no pudieron confirmar otros resultados. La mitad de las familias presentan ligamiento en los genes que codifican la hamartina y la otra mitad de las familias en los genes que codifican la tuberina. Tan sólo en el 10% de los casos no existe ligamiento, lo cual parece deberse a errores de tipificación genotípica10. La gran variabilidad en la expresión clínica de la esclerosis tuberosa entre los miembros de una misma familia se explica por la necesidad de una segunda mutación en el par homólogo para que la enfermedad se manifieste5, 6. El índice de nuevas mutaciones es muy elevado (50% a 75% de todos los casos), y como en nuestro estudio, se han descrito casos de familias con padres aparentemente normales y uno o varios hijos afectados. Un mosaicismo somático puede explicar la variable expresión intrafamiliar y la aparente falta de penetrancia de la enfermedad. De hecho se han descrito numerosos casos de discordancia genética, incluso entre gemelos idénticos11. Estos casos, sin embargo, no son muy frecuentes, aunque su existencia se revela importante si no crucial en el estudio de la fisiopatología de esta enfermedad. Los diferentes autores consideran diversas hipótesis para explicar este fenómeno:
-- Influencia de factores medioambientales.
-- Efectos de orden aleatorio de manera análoga a lo que sucede en el modelo genético del retinoblastoma.
-- Que las mutaciones en el gen casual de la esclerosis tuberosa sucedan tras la división del cigoto inicial en dos cigotos que evolucionen hacia dos embriones diferentes.
-- Que la versatilidad intergemelar sea debida a un fenómeno de imprintting12.
Schnur13 en 1995 ya dejó en el aire algunas preguntas que comienzan a contestarse en la actualidad: ¿hay otros genes que interactúen y puedan modificar la progresión de la enfermedad?, ¿hay genes adicionales que pueden determinar una especial susceptibilidad individual para experimentar una segunda mutación?, ¿es necesaria una terapia mucho más directa para prevenir las serias complicaciones congénitas asociadas a la esclerosis tuberosa y evitar su progresión posterior? Por ello, para diagnosticar de una manera definitiva esta enfermedad habrá que recurrir a la identificación de la mutación genética de la esclerosis tuberosa en sangre periférica, fibroblastos o hamartomas.
MANIFESTACIONES CLINICAS
El cuadro clínico se caracteriza por la aparición de múltiples tumores benignos (hamartomas) y malformaciones y anomalías (hamartrias; tabla 1) que afectan a la mayoría de los órganos y configuran un complejo conjunto de criterios diagnósticos (tabla 2) que de manera periódica son revisados por la National Tuberous Sclerosis Association (NTSA), lo que supone alejarnos de la tríada que clínicamente ha definido a esta entidad durante tantos años. Algunas formas escapan al desarrollo polivisceral y permanecen estáticas, localizadas en uno o varios órganos y determinan las formas fustres o incompletas14.


Manifestaciones dermatológicas
Las manifestaciones dermatológicas son las más fáciles y menos invasivas para llegar al diagnóstico.
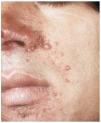
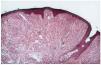
--Los mal llamados adenomas sebáceos de Pringle (fig. 1) están presentes en el 83% al 90% de los casos. Son patognomónicos y se localizan de forma preferente en surcos nasolabiales, mejillas, mentón, cuero cabelludo, frente y más raramente en orejas. Rara vez aparecen en el nacimiento y suelen hacerlo entre los 3-10 años y se estabilizan en la adolescencia para persistir de por vida. En raras ocasiones pueden formar masas fungosas de mayor tamaño. Histológicamente son angiofibromas faciales con intensa fibrosis dérmica y dilatación de los capilares. La morfología estrellada de los fibroblastos le confieren un aspecto glial. Existe proliferación perivascular y perifolicular de fibroblastos con disminución y/o desaparición de fibras elásticas (fig. 2).
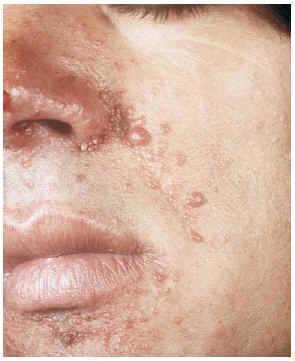
FIG. 1.--Fibroangiomas múltiples en mejillas.
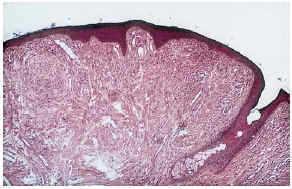
FIG. 2.--Angiofibroma: dilatación capilar y fibrosis dérmica.
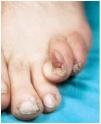
-- Los tumores de Koenen (fig. 3) son fibromas periungueales o subungueales rosados y más o menos alargados de 5-10 mm de longitud que pueden llegar a destruir el lecho ungueal. Aparecen en la pubertad y continúan con el desarrollo. A veces coexisten varios en la misma uña.
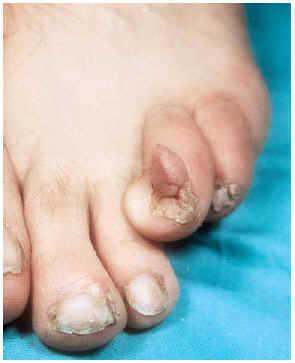
FIG. 3.--Tumor de Koenen en periniquio.
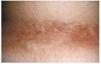
-- En la región lumbosacra se sitúan de forma característica las placas chagrin (fig. 4), carac- terizadas por ser áreas de piel ligeramente elevada, amarillenta, de tacto similar a la lija, cuyo tamaño oscila entre 1 y 5 cm de diámetro y que en ocasiones se rodean de otras más pequeñas. La histología muestra la disposición esclerótica del colágeno en dermis profunda de manera similar a la morfea. Suele existir disminución y fragmentación de fibras elásticas.
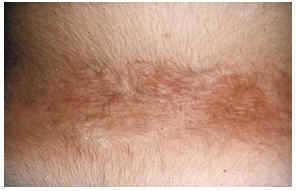
FIG. 4.--Placa chagrin en zona lumbar.
-- Sin embargo, para un precoz diagnóstico de la enfermedad son de extraordinaria importancia las manchas acrómicas congénitas o de aparición en los primeros meses de vida. Están presentes en el 85% de los casos y junto con la presencia de convulsiones no febriles en la infancia son dos datos clave para establecer un diagnóstico de sospecha sólido. Son de aspecto oval o lanceolado (fig. 5), de 1-3 cm de longitud y con morfología en hoja de fresno tal y como describen los textos clásicos, situándose sobre todo en tronco y extremidades15. En muchos casos es necesaria una exploración con luz de Wood para su observación. Desde el punto de vista histopatológico se caracteriza por presentar un número normal de melanocitos con atenuación de la doparreacción (fig. 6). Con microscopía electrónica se aprecia una disminución de función y tamaño de los melanosomas.
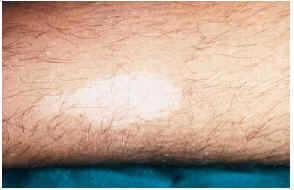
FIG. 5.--Manchas acrómicas en pierna.
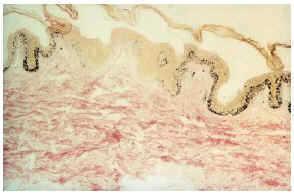
FIG. 6.--Aspecto histológico de una mancha acrómica (Fontana).
-- Con menos frecuencia, pero también presentes, se hallan fibromas molluscum, hiperplasia gingival, angiomas, manchas de café con leche, poliosis, onicolisis, pits dentales, etc.
Manifestaciones neurológicas
Las principales alteraciones neurológicas de la esclerosis tuberosa son las convulsiones tónico-clónicas (las padecen el 90% de los pacientes), el retraso mental y las alteraciones de la conducta. Estas convulsiones pueden adoptar la forma de crisis mioclónicas, espasmos infantiles (hipsarritmias de West) o en adultos crisis generalizadas tónico-clónicas o parciales16. La aparición de túberes corticales está en estrecha relación con el pronóstico neurológico del paciente y son los principales responsables de los episodios convulsivos17. Parece que la evolución favorable, tanto de las funciones neuropsíquicas como de las crisis epilépticas está relacionada con menor número de túberes que en los sujetos más retrasados y con crisis de difícil control18. Al comprobar que el pronóstico depende del adecuado control de las convulsiones, no deben ahorrarse medios para conseguirlo. Incluso si las convulsiones provienen de un nódulo identificable debe procederse a cirugía de la epilepsia, especialmente si el túber identificado como responsable de las crisis es fácilmente accesible a la cirugía. Sin embargo, no hay relación entre la frecuencia de las crisis, anormalidades cutáneas y el retraso mental. Otras manifestaciones son ataxia, oftalmoplejía, parálisis espásticas, hemianopsias, etc.
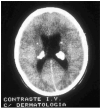
Las manifestaciones tumorales son hamartomas tuberosos múltiples, bien delimitados, con potencial blastomatoso, benigno o maligno que se encuentran a nivel de corteza cerebral y más raramente en cerebelo o en médula espinal. Su transformación maligna puede conducir a astrocitomas de células gigantes y glioblastomas con sintomatología de expansión intracraneal: hipertensión, cefaleas, vómitos, alteraciones visuales y papiledema. El electroencefalograma (EEG) está alterado en el 80% de los pacientes, pero las anomalías encontradas no son específicas de la enfermedad. El líquido cefalorraquídeo (LCR) es normal o, como mucho, puede presentar una ligera elevación de las proteínas. En los estudios radiológicos simples se pueden observar calcificaciones de los ganglios de la base o paraventriculares (fig. 7) y en la TAC o en la RMN cerebral tubérculos corticales, nódulos subependimarios, lesiones en la materia blanca periventricular profunda, etc.

FIG. 7.--Calcificaciones paraventriculares (tercer ventrículo).
Manifestaciones oftalmológicas
Los signos oculares son fortuitos y se descubren con una frecuencia del 50% durante la consulta oftalmológica rutinaria, pues al ser asintomáticos pasan inadvertidos, sobre todo si no existe un componente extraocular orientativo. Su lesión característica es el hamartoma astrocítico del nervio óptico (fig. 6). Tiene escasa tendencia a crecer aunque sí se calcifica formando drusas. El sangrado de los vasos anómalos del tumor determina hemorragia vítrea19. La angiografía fluoresceínica permite observar en el tiempo arterial el llenado rápido de los vasos dilatados intratumorales, y en la fase venosa la existencia de capilares tortuosos y dilatados. En ocasiones los hamartomas retinianos surgen en la periferia como manchas grises, ovaladas, algo prominentes, semejando fibras de mielina o como formaciones quísticas libres, aunque tanto unas como otras no son específicas de la enfermedad. Respecto a ellos es necesaria una observación minuciosa, así como controles periódicos, por la elevada tendencia a la mortalidad debida a la aparición de complicaciones generales cuya incidencia se ve incrementada después de los 30 años de edad.
Manifestaciones renales
En la esclerosis tuberosa se pueden hallar quistes renales bilaterales, angiomiolipomas y en ocasiones oncocitomas renales20. El angiomiolipoma renal es un tumor de rara observación en la población general, si bien casi el 50% de los casos aparecen en pacientes con esclerosis tuberosa. Son únicos y más frecuentes en el sexo femenino. Clínicamente son asintomáticos, pero a veces se manifiestan como masa abdominal palpable, dolor en zona lumbar y más raramente como hematuria, uremia o albuminuria. La incidencia de insuficiencia renal crónica es baja y el fallecimiento del paciente por causa renal es debido al desarrollo de una enfermedad poliquística y requiere diagnóstico diferencial con otras neoplasias renales.
Otras manifestaciones
La clínica de la esclerosis tuberosa se completa con manifestacions óseas21 (lesiones quísticas en falanges, matacarpo y metatarso, lesiones osteoescleróticas en columna (fig. 7), pelvis y cráneo, formación de nuevo hueso perióstico, osteoartropatía hipertrófica, gigantismos monomiélicos, etc.), cardíacos22 (rabdomioma cardíaco, mucho más común en niños que en adultos; comunicaciones intraventriculares, aorta bicuspídea, fibroelastosis endomiocárdica, etc.), pulmona-res23, 24. Por otro lado son raras, ya que se presentanen menos del 1% de los casos: manifestaciones pleurales, fibrosis pulmonar, se manifiesta clínicamente por medio de tos seca, disnea, hemoptisis y neumotórax espontáneo, asociándose los casos de mortalidad a la aparición de insuficiencia respiratoria aguda y/o cor pulmonale); poliposis intestinal, afectación hepática, esplénica, pancreática, hamartomas en tiroides, suprarrenales y trastornos endocrinometabólicos, del tipo pubertad precoz y malformaciones de diferente índole y consideración15, 25, son otras manifestaciones a tener en cuenta.
DIAGNOSTICO
Una vez realizado el diagnóstico clínico en virtud a los criterios clínicos ya expuestos, mencionamos a continuación las pruebas complementarias para evaluar a un paciente diagnosticado de novo de esclerosis tuberosa, según las recomendaciones efectuadas en el documento de consenso elaborado por Roach26 et al en 1999:
-- TAC y RMN craneal 27. Son imprescindibles para evaluar disfunción neurológica y la presencia simultánea de astrocitoma glial subependima-rio, muchas veces presente en el momento del diagnótico inicial. Deberá ser reevaluado cada 1-3 años.
-- Electroencefalograma. Sólo tiene utilidad manifiesta cuando el paciente ha comenzado su cuadro clínico inicial con crisis epilépticas.
-- Ecografía renal. Debe realizarse en el momento del diagnóstico para evaluar la presencia de angiomiolipomas28, tumores renales y/o riñones poliquísticos. Debe repetirse con una frecuencia que oscila entre 1 y 3 años.
-- Electrocardiograma. Debe realizarse en el momento del diagnóstico dada la probabilidad de desarrollar arritmias que presentan estos pacientes. El síndrome de Wolff-Parkinson-White es el más frecuente y suele presentarse tras el inicio del tratamiento con carbamacepina para el tratamiento de crisis epilépticas29.
-- Ecocardiograma. Se recomienda realizar ecocardiograma sólo en aquellos pacientes que muestren síntomas compatibles con rabdomioma cardíaco, puesto que las arritmias de mayor incidencia no se detectan con este método, sino mediante electrocardiograma (ECG) y las posibilidades de desarrollar disfunción cardiológica disminuyen con el crecimiento del paciente si en principio no ha presentado sintomatología adicional.
-- Examen oftalmológico. Es obligado realizar fondo de ojo en el momento del diagnóstico de estos pacientes para examinar la aparición de maculopatía asociada que puede condicionar el pronóstico y calidad de vida de estos pacientes.
-- Examen dermatológico. El examen dermatológico, también fundamental, es especialmente útil cuando las manifestaciones cutáneas son atípicas o cuando el diagnóstico de esclerosis tuberosa no es claro y deberemos incidir en el uso de la luz de Wood para el examen de lesiones hipocrómicas.
-- Pruebas psicomotrices y de desarrollo neurológico30. El panel de recomendaciones al inicio recomienda un screening de las características psicomotrices, neurológicas y comportamentales del paciente, el cual debe ser repetido en el caso de los niños en el momento de inicio de escolarización y en caso de detectar posibles anomalías a lo largo de su proceso educacional.
-- Diagnóstico molecular. Las pruebas de diagnóstico molecular no están disponibles de manera rutinaria en la mayoría de los hospitales, pero en el momento que puedan llegar a estarlo la caracterización de los defectos génicos podría llegar a identificar un menor o mayor riesgo de desarrollo de las diferentes complicaciones. En un futuro será un método válido para determinar qué fenotipos se correlacionan con uno u otro defecto genético y de esta manera establecer un pronóstico acertado en el diagnóstico inicial.
Como apreciamos, el protocolo que es preciso aplicar para estudiar al paciente es amplio y queda recogido en la tabla 3, en la cual completamos los criterios referidos por Naranjo y Jiménez-Devesa15 con los recientemente incorporados por Roach en la última Conferencia de Consenso referida al tema que nos ocupa.

PRONOSTICO Y TRATAMIENTO
La supervivencia de un paciente con esclerosis tuberosa está ligada a la gravedad de las manifestaciones neurológicas de la enfermedad, y en su defecto al desarrollo de una insuficiencia renal crónica debido a la sustitución de tejido renal parenquimatoso por hamartomas o la aparición de neoplasias malignas. No existe tratamiento específico. La clínica neurológica se controlará de forma sintomática con farmacología anticomicial y en los casos de hipsarritmia de West con ACTH (adrenocorticotropina).
En conclusión, la esclerosis tuberosa o enfermedad de Pringle Bourneville es una facomatosis cuyas manifestaciones clínicas son extremadamente variables y ante cuya sospecha es indispensable aplicar un complicado pero necesario protocolo diagnóstico, que necesita colaboración interdisciplinar para facilitar el mejor trato y la mejor calidad de vida al paciente.


