
INTRODUCCION
El escleromixedema, también conocido como síndrome de Arndt-Gottron, es una enfermedad rara, de etiología desconocida, caracterizada por una erupción simétrica y extensa de pápulas firmes, céreas, con marcada esclerosis e induración de la piel1-4. Se localiza principalmente en cara, cuello, parte superior del tronco, manos y antebrazos. En la mayoría de los casos se asocia con gammapatía monoclonal y trastornos sistémicos2-6. Histológicamente se observan depósitos de mucina en dermis reticular superior y media, aumento de colágeno y proliferación de fibroblastos irregularmente distribuidos2-4. Gran parte de los pacientes descritos en la literatura médica bajo la denominación de liquen mixedematoso o mucinosis papulosa, sin indicación de subtipo, corresponden en realidad a escleromixedema con gammapatía monoclonal4. Se describe un nuevo caso de escleromixedema con importantes manifestaciones cutáneas y sistémicas que hemos tenido oportunidad de estudiar su evolución en nuestro servicio durante un período de 15 años. Durante este tiempo ha recibido distintos tratamientos, respondiendo únicamente al clorambucilo y posteriormente a la asociación de ciclos de melfalán con prednisona.
CASO CLINICO
Una mujer de 55 años, con antecedentes personales de hipertensión arterial y meningioma cerebral presentaba desde 1988, pápulas liquenoides junto con edema y endurecimiento en cara, pabellones auriculares, parte superior del tronco, antebrazos, dorso de manos, rodillas y pies. Estas lesiones experimentaron períodos de mejoría y remisión parcial que la paciente relacionaba con un tratamiento recibido entre 1992 a 1998. En los últimos 4 años se produjo un empeoramiento progresivo, con aparición creciente de nuevas pápulas sobre un fondo de eritema e induración. Como sintomatología sistémica asociada refería adelgazamiento, astenia, disfagia y odinofagia con regurgitación ocasional de alimentos, artralgias, debilidad de cintura escapular y pelviana con dificultad para caminar y levantarse a partir del decúbito, mareos ocasionales y fenómeno de Raynaud.
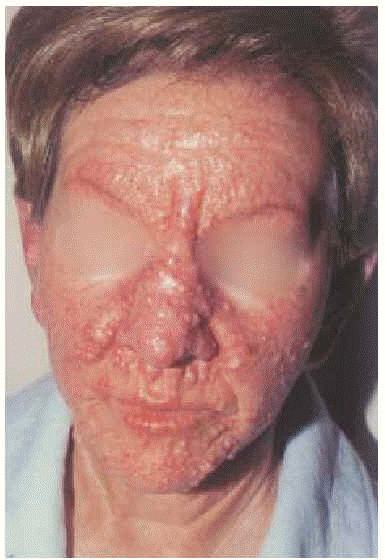
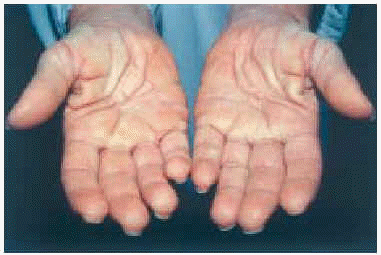
En la exploración cutánea se observaba una erupción simétrica y extensa de pápulas firmes, céreas, de 2-10 mm de diámetro, localizadas en cara, pabellones auriculares, región submamaria, superficie de extensión de antebrazos, muñecas, dorso de manos y dedos (fig. 1). En general, estas pápulas se distribuían de modo lineal. La piel mostraba aspecto brillante, algo eritematosa, con edema y sensación de endurecimiento al tacto. En la glabela se observaba un surco longitudinal profundo que daba una imagen a la cara de facies leonina. En las articulaciones interfalángicas proximales se apreciaba un borde sobreelevado con depresión central (signo del buñuelo). El engrosamiento de partes blandas en las palmas de las manos hacia que éstas mostrasen un aspecto escrotal (fig. 2). Los dedos de las manos presentaban un ligero grado de esclerodactilia, sin telangiectasias ni calcinosis, y en las extremidades, se apreciaba livedo reticularis.
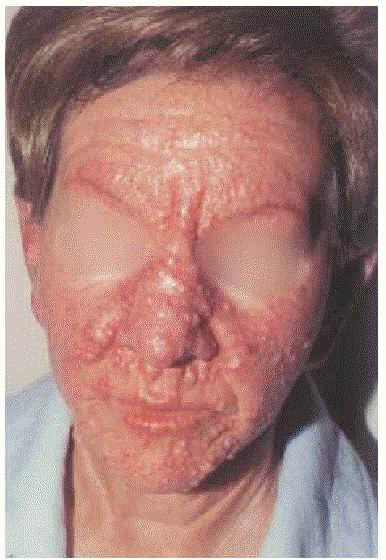
Fig. 1.--Múltiples pápulas blanquecinas, algunas de disposición lineal, sobre una piel eritematosa y brillante.
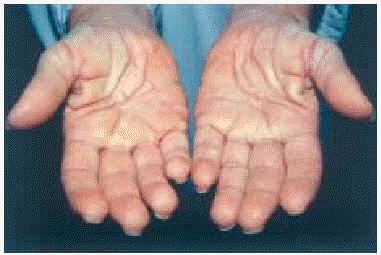
Fig. 2.--Engrosamiento de partes blandas en palmas de manos.
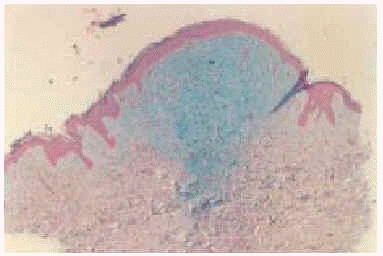
El estudio al microscopio óptico de la piel con hematoxilina-eosina mostró depósitos de mucina en dermis reticular superior y media, junto con esclerosis y proliferación de fibroblastos con aspecto alargado o estrellado. Las tinciones especiales de azul alcián y hierro coloidal mostraron positividad para mucina (fig. 3).
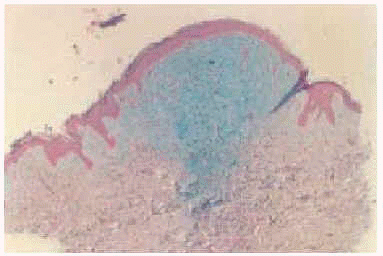
Fig. 3.--Tinción con azul alcián. Depósitos de mucina en dermis reticular superior y media.
Los análisis de laboratorio efectuados incluyeron: hemograma, velocidad de sedimentación globular (VSG), estudio de coagulación, sistemático y sedimento de orina, bioquímica hemática (glucosa, urea, creatinina, GOT, GPT, GGT, bilirrubina, colesterol, triglicéridos, ácido úrico, hierro, CPK, LDH), proteinograma, determinación de inmunoglobulinas, autoanticuerpos (ANA, ENA, Anti-DNA, anticentrómero, anti-scl 70, antimitocondriales, antimúsculo liso y antigliadina), fracciones C3 y C4 del complemento, estudio de tiroides (TSH, T3 y T4, anti-TPO), proteína de Bence-Jones en orina y serología hepatitis B y C. Los resultados de todos ellos fueron normales o negativos excepto hemoglobina de 9,4 g/l, ligera leucocitosis (10.100 μl) con eosinofilia (18 %), VSG de 87 mm a la primera hora, CPK 162 U/l (normal < 150 U/l), banda monoclonal IgGλ de 10,9 g/l y gammapatía policlonal de 6,2 g/l. La biopsia de médula ósea fue normal.
Entre las exploraciones complementarias realizadas destacamos un electromiograma con perfil parcialmente miogénico. El electrocardiograma, radiografía de tórax y las pruebas de función respiratoria y ventilatoria pulmonar fueron normales. La tomografía computarizada (TC) cerebral presentaba las imágenes características del meningioma en el borde superior de clinoides anterior derecha.
La paciente ha recibido a lo largo de su proceso diferentes tratamientos. Al inicio de su enfermedad se trató con cloroquina, PUVA y retinoides orales sin respuesta satisfactoria. Desde 1992 a 1998 fue tratada con clorambucilo en dosis y pautas variables (se inició con 4 mg/día los primeros meses, para después reducir de manera progresiva hasta 4 mg al mes durante el último año) que dieron lugar a una mejoría importante de su cuadro los primeros años de tratamiento para finalmente mostrarse refractario a éste cuando se reintrodujo en el año 2001. En abril de 2002 comenzó un tratamiento con ciclos de 4 días al mes de melfalán 10 mg/día y prednisona 40 mg/día con lo que mejoraron parcialmente las lesiones cutáneas y remitió totalmente la sintomatología sistémica. La paraproteinemia monoclonal se redujo a 4,2 g/l.
DISCUSION
Nuestra paciente presentaba una sintomatología cutánea característica e intensa en forma de pápulas liquenoides que asentaban sobre un piel indurada en las localizaciones típicas. Estas lesiones, en su conjunto, progresaron con los años y dieron lugar a una importante desfiguración de la cara. No obstante, queremos reseñar que las pápulas que en su inicio estaban localizadas en rodillas y pies remitieron con el tratamiento de clorambucilo y no reaparecieron de nuevo. También pudimos observar cómo las pápulas liquenoides del dorso de manos, muy manifiestas al comienzo de la enfermedad, aparecían más tenues cuando el escleromixedema estaba más evolucionado. Además de las lesiones cutáneas, la paciente presentaba importante afectación sistémica destacando principalmente la sintomatología digestiva (disfagia, odinofagia, regurgitación) y miopatía que la impedía realizar con normalidad ciertas actividades cotidianas básicas (caminar, incorporarse desde la posición de decúbito, etc.). Casos graves como el de nuestra paciente suponen un importante reto terapéutico.
El tratamiento del escleromixedema no está estandarizado y la elección terapéutica es con frecuencia empírica7. Entre los múltiples tratamientos utilizados están los corticoides tópicos y sistémicos2,4, PUVA8, retinoides orales9,10, fotoquimioterapia extracorpórea y bolos de corticoides7, ciclosporina11, interferón alfa12 y trasplante autólogo de células madre13. La presencia de gammapatía monoclonal y la posibilidad de evolución hacia mieloma ha conducido a la utilización de monoquimioterapia7, principalmente con clorambucilo14, ciclofosfamida6,15 y sobre todo melfalán2-4,16-18. Este último es el que con mayor frecuencia se ha propuesto como tratamiento en escleromixedema. Las pautas y formas de administración del melfalán son variables y oscilan entre dosis bajas (2-4 mg/día), y mantenidas durante varios meses y/o años, a aquellos casos en los que se administra en forma de ciclos combinando 10 mg de melfalán con 15 mg de prednisona3. Los resultados, en general, han sido buenos con resolución parcial o total de las lesiones cutáneas en la mayoría de los casos; sin embargo, esta terapia no está exenta de efectos secundarios importantes, en particular su implicación en el desarrollo de sepsis o neoplasias hematológicas. Gabriel et al2 observaron regresión de las lesiones cutáneas en 8 de 12 pacientes tratados con melfalán; a pesar de ello, 5 de 11 (45 %) desarrollaron complicaciones graves, ya que dos murieron de sepsis secundaria a citopenia y tres desarrollaron enfermedad hematológica (dos leucemia, uno linfoma). Dinneen y Dicken3 también encontraron una importante mortalidad en relación con el melfalán, ya que de 17 pacientes tratados, 6 muertes se relacionaron directamente con la terapia. Estos resultados han llevado a que algunos autores, como Rongioletti y Rebora4 recomienden su uso únicamente para aquellos pacientes que estén desfigurados, inválidos o tengan complicaciones sistémicas. En nuestro caso, dado el carácter incapacitante de la enfermedad y la deformidad que producía optamos por pautar la asociación de prednisona y melfalán, logrando una remisión de la sintomatología sistémica y mejoría de las lesiones cutáneas, con reducción considerable del valor de la paraproteína monoclonal IgGl.


