








La epidermólisis ampollosa (EA) engloba un grupo de enfermedades hereditarias que afectan a uno de cada 17.000 nacidos vivos en el mundo. Consiste en la formación de ampollas ante el menor traumatismo que afectan a la piel y a las mucosas. Esta enfermedad empeora seriamente la calidad de vida. El diagnóstico se realiza principalmente por mapeo por inmunofluorescencia y microscopía electrónica. El tratamiento es sintomático, aunque se están investigando nuevas terapias celulares y moleculares. Esta revisión presentará información relevante sobre la biología molecular, la sintomatología clínica, el diagnóstico y el tratamiento de la EA, con la clara intención de proporcionar un mejor cuidado a los pacientes que padecen esta enfermedad.
Epidermolysis bullosa is a group of hereditary diseases affecting 1 in 17 000 live births worldwide. It consists of blistering of the skin and mucous membranes in response to minimal trauma. The disorder seriously affects the patient's quality of life. Diagnosis is based on immunofluorescence mapping and electron microscopy. Treatment is symptomatic, although new cellular and molecular therapies are currently under investigation. This review covers aspects of the molecular biology, clinical presentation, diagnosis, and treatment of epidermolysis bullosa relevant to improving the care for affected patients.
Las epidermólisis ampollosas (EA) representan un grupo heterogéneo de patologías hereditarias caracterizadas por una marcada fragilidad de la piel y las mucosas, que desencadena la formación de ampollas y úlceras en respuesta a traumatismos menores1. Los sitios que más se afectan son los expuestos a la fricción y la presión frecuentes, razón por la que se les ha denominado enfermedades mecano-ampollosas 2. Las manifestaciones extracutáneas, como la afección de dientes y anejos cutáneos, así como de los epitelios gastrointestinal, vesicourinario y pulmonar, aumentan la complejidad clínica de esta patología, hasta la fecha incurable.
La incidencia reportada de este padecimiento varía por zonas geográficas, afectando aproximadamente a uno de cada 17.000 nacidos vivos y con una estimación mundial de 500.000 casos actuales3,4. Sin embargo, en muchos países como en México no se conoce por completo el porcentaje de niños que nacen con EA 2. Esta enfermedad no muestra predilección por raza ni etnia1 y afecta a ambos sexos por igual. Se han identificado más de 10 genes involucrados en la etiología de la EA, documentándose más de 1.000 mutaciones que pueden ocurrir de novo, o seguir un patrón de herencia autosómica dominante o recesiva5. Existe también una forma adquirida de EA que se presenta en la cuarta o quinta década de la vida y se debe a la producción de autoanticuerpos IgG contra el colágeno VII6.
Existen tres formas principales de la enfermedad: EA simple (EAS), de la unión (EAU) y distrófica (EAD). La EAS es la presentación más frecuente (92 %), seguida por la EAD (5 %) y finalmente por la EAU (1 %)7. Se diferencian entre sí de acuerdo al sitio de separación y formación de las ampollas en la piel, ya sea superficial a la membrana basal epidérmica, dentro de ella o por debajo de la misma8.
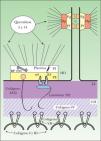
DermatopatologíaLa epidermis, un tejido completamente celular, se compone de múltiples capas de queratinocitos en varias etapas de diferenciación, desde una capa basal de queratinocitos hasta una capa superficial de células córneas. La dermis, en cambio, consiste en un conjunto de células dispersas en una abundante matriz extracelular9. La membrana basal epidérmica, localizada en la unión dermo-epidérmica, cumple con la función de mantener la adhesión entre estos dos tejidos estructuralmente diferentes mediante una compleja red de moléculas de adhesión finamente relacionadas entre sí10 (fig. 1).
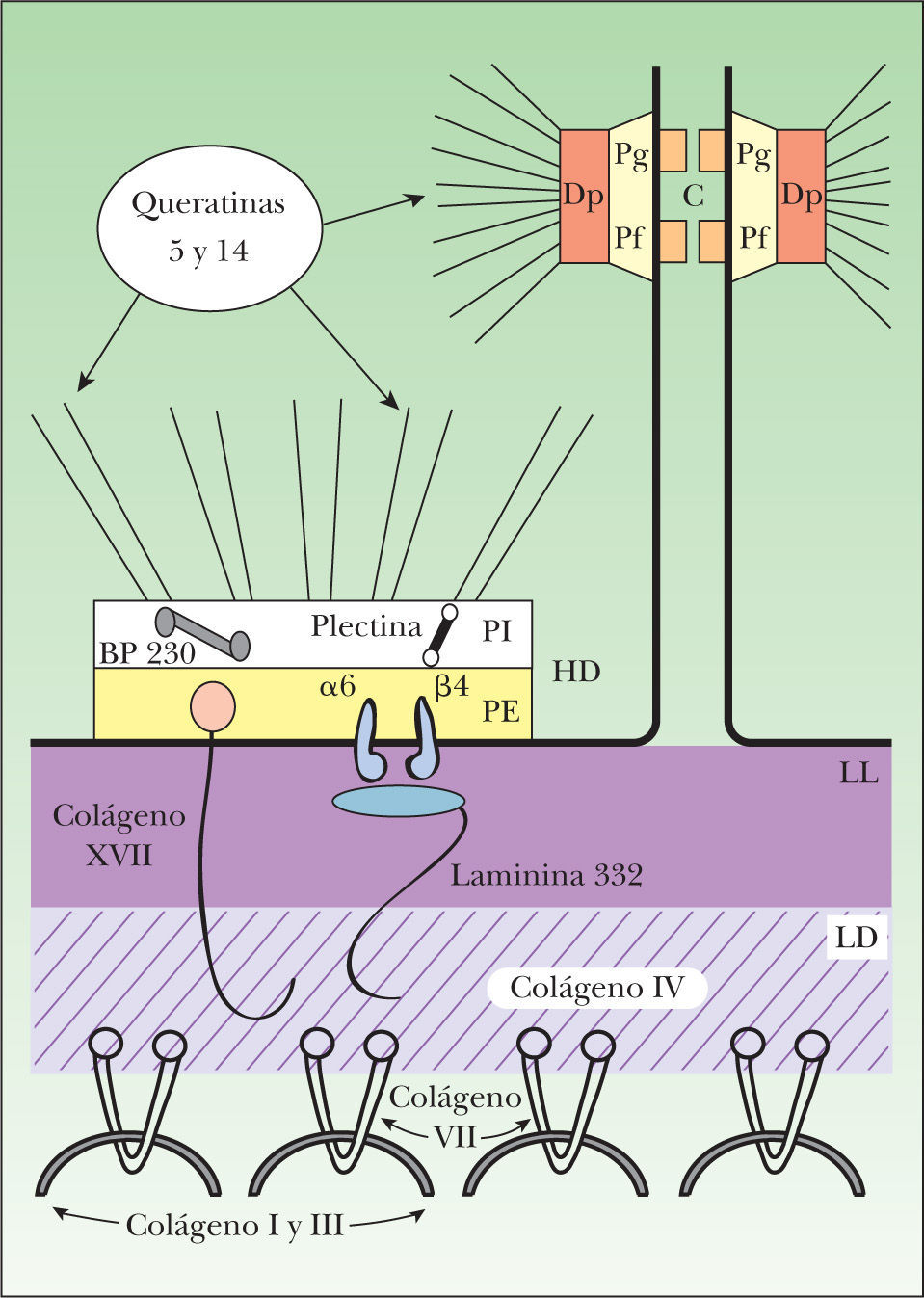
Organización molecular de la membrana basal epidérmica. Los filamentos intermedios, compuestos por las queratinas 5 y 14, se encuentran en el citoplasma de los queratinocitos basales y se unen en la placa interna de los hemidesmosomas con la plectina y el BP230. Estas dos plaquinas interactúan con dos moléculas transmembranales, la integrina α6β4 y el colágeno XVII. La integrina α6β4 es el receptor para el ligando extracelular de la laminina 332, que se une a su vez con el colágeno VII a través del colágeno IV de la lámina densa. El colágeno VII es el principal componente de las fibrillas de anclaje e interactúa también con el colágeno XVII. α6: subunidad alfa de la integrina α6β4; β4: subunidad beta de la integrina α6β4; BP230: antígeno del penfigoide ampolloso de 230kDa; C: cadherinas; Dp: desmoplaquina; HD: hemidesmosoma; LD: lámina densa; LL: lámina lúcida; PE: placa externa del hemidesmosoma; Pf: placofilina; Pg: placoglobina; PI: placa interna del hemidesmosoma.
El citoesqueleto de los queratinocitos basales está constituido por una red citoplásmica de filamentos intermedios, principalmente de queratina 5 y queratina 14, que participan en la coordinación de la forma celular y en la integridad estructural de la epidermis mediante su conexión a los desmosomas y hemidesmosomas. Las células epidérmicas se comunican entre sí por medio de desmosomas y puentes de unión intercelulares. Los desmosomas son complejos especializados que forman uniones intercelulares estrechas entre células epiteliales adyacentes. Sus principales componentes son las caderinas desmocolina y desmogleína, la placoglobina, la placofilina y la desmoplaquina.
Mediante el estudio de microscopía electrónica se observa que la membrana basal epidérmica se divide en tres áreas: los hemidesmosomas, la lámina lúcida y la lámina densa. Los hemidesmosomas constituyen la unidad principal de adhesión en la unión dermo-epidérmica de la piel. Se componen de una placa interna citoplásmica y una placa externa en continuidad con la membrana celular. Los filamentos de queratina se insertan en la placa interna de los hemidesmosomas mediante su interacción con dos proteínas de la familia de las plaquinas: la plectina y el antígeno de penfigoide ampolloso de 230kDa (BP230 o BPAG1).
Las plaquinas se asocian a su vez con dos proteínas transmembranales contenidas en la placa externa de los hemidesmosomas: la α6β4 integrina y el antígeno de penfigoide ampolloso de 180kDa (BP180 o BPAG2), también conocido como colágeno XVII. El dominio extracelular de la a6b4 integrina se extiende hasta la lámina lúcida, donde se une con la laminina 332 (antes conocida como laminina 5). El dominio extracelular del colágeno XVII se extiende hasta la lámina densa. La laminina 332 y el colágeno XVII constituyen los filamentos de anclaje que unen los filamentos intermedios con las fibrillas de anclaje de la dermis papilar.
El principal componente de las fibrillas de anclaje es el colágeno VII. Esta proteína forma dímeros que se asocian lateralmente en haces semicirculares, y cuyos extremos se insertan en la lámina densa, donde interactúan con los filamentos de anclaje por medio de su asociación con el colágeno IV. Los haces semicirculares del colágeno VII forman una especie de bandas que atrapan a las proteínas de la matriz dérmica, como el colágeno I y tipo III, manteniéndolas unidas a la membrana basal epidérmica9–13.
Alteraciones moleculares y genéticasLa presencia de defectos genéticos que alteren una o más moléculas esenciales de la unión dermo-epidérmica conduce a una pérdida de la capacidad de adhesión de la membrana basal epidérmica 14 (tabla 1). La gravedad de las manifestaciones clínicas varía mucho entre los tipos y subtipos de EA, abarcando un espectro que va desde la formación de ampollas pequeñas hasta la presencia de ampollas y erosiones extensas, cicatrización grave y complicaciones mortales. No es posible adjudicar estas diferencias a la presencia de un gen específico anormal, ya que una misma anormalidad genética puede asociarse con manifestaciones clínicas sustancialmente diferentes.
Alteraciones genéticas en la epidermólisis ampollosa
| Tipo de epidermólisis ampollosa (EA) | Gen afectado | Proteína que codifica |
| EA simple | PKP1 | Placofilina 1 |
| DSP | Desmoplaquina | |
| KRT5 | Queratina 5 | |
| KRT14 | Queratina 14 | |
| PLEC1 | Plectina | |
| ITGA6, ITGB4 | Integrina α6β4 | |
| EA de la unión | LAMA3, LAMB3, BLAMC2 | Laminina 332 |
| COL17A1 | Colágeno XVII | |
| ITGA6, ITGB4 | Integrina α6β4 | |
| EA distrófica | COL7A1 | Colágeno VII |
| Síndrome de Kindler | KIND1 | Kindlina 1 |
En las últimas décadas el avance en las técnicas de análisis molecular y genético ha permitido evaluar las mutaciones específicas presentes en pacientes con EA, encontrándose diferentes tipos de mutaciones (deleciones, inserciones, con o sin sentido, de corte y empalme, deleción/inserción sin lectura del marco genético) en los genes de las moléculas de adhesión de la membrana basal epidérmica. Inclusive en muchos casos de EA con herencia autosómica recesiva se observan dos mutaciones diferentes en el mismo individuo (heterocigosidad compuesta). Los tipos y combinaciones de las mutaciones presentes, así como sus consecuencias en la transcripción al ARN mensajero y traducción a proteínas, determinan el tipo y la gravedad de la EA en cada paciente. Esta variabilidad fenotípica explica el espectro de las manifestaciones clínicas observadas15–21.
ClasificaciónEn 1962 Pearson desarrolló el primer sistema de clasificación para la EA. Sirviéndose del microscopio electrónico definió los tres tipos principales de EA, para lo que se basó en el plano de despegamiento5. Esta clasificación se ha utilizado hasta ahora en la práctica clínica, en la enseñanza y en la investigación.
Durante la década de los ochenta, con el desarrollo de la tecnología de inmunofluorescencia, se hizo uso de anticuerpos monoclonales y policlonales para la inmunotinción de muestras de piel, demostrándose que varios subtipos de EA podían distinguirse por diferencias en los patrones de tinción de antígenos. En el año 1988 se realizó la primera reunión de expertos en la ciudad de Washington DC, para intentar establecer un consenso internacional en la clasificación de la EA, tomando ventaja de los datos que habían sido generados en los Estados Unidos por el National Epidermolysis Bullosa Registry. El desarrollo de las técnicas de análisis de mutaciones permitió conocer con exactitud los defectos moleculares y genéticos de los distintos subtipos de EA. En 1999 se realizó el segundo consenso internacional en Chicago, en el que se tomaron en cuenta nuevas entidades clínicas descritas y los resultados de estudios de análisis de mutaciones.
Durante los últimos años se ha aprendido mucho sobre el espectro de la EA, tanto en el ámbito clínico como molecular. A la luz de los nuevos conocimientos se consideró necesario realizar una nueva revisión del sistema de clasificación de la EA. En mayo de 2007 las autoridades mundiales en el campo de la EA se reunieron en Viena, elaborando el Tercer Consenso Internacional sobre Diagnóstico y Clasificación de Epidermólisis Ampollosa. En este último consenso se han incluido otras entidades clínicas en el espectro de la EA, basándose en el hecho de que comparten características con los tipos bien establecidos de EA y que su transmisión obedece a un patrón hereditario. Entre las nuevas variantes se incluyen el síndrome de Kindler, el síndrome laringo-onico-cutáneo (LOC), la EAS letal acantolítica, la EAS circinada migratoria y la deficiencia de placofilina. Se ha eliminado el término«EA hemidesmosomal», y se ha cambiado el nombre de algunos subtipos previamente identificados por su epónimo a un nombre clínicamente descriptivo 5 (tabla 2).
Clasificación de acuerdo al Tercer Consenso Internacional sobre Diagnóstico y Clasificación de la Epidermólisis Ampollosa
| Tipo | Subtipo mayor | Subtipo |
| Epidermólisis ampollosa simple (EAS) | Suprabasal | Letal acantolítica |
| Deficiencia de placofilina | ||
| EAS superficialis (EASS) | ||
| Basal | EAS, localizada (EAS-loc)* | |
| EAS, Downling-Meara (EAS-DM) | ||
| EAS, otras generalizadas (EAS, gen-nonDM; EAS, gen-nDM)** | ||
| EAS con distrofia muscular (EAS-MD) | ||
| EAS con pigmentación moteada (EAS-MP) | ||
| EAS con atresia pilórica (EAS-PA) | ||
| EAS, autosómica recesiva (EAS-AR) | ||
| EAS, Ogna (EAS-Og) | ||
| EAS, circinada migratoria (EAS-migr) | ||
| Epidermólisis ampollosa de la unión (EAU) | EAU, Herlitz (EAU-H) | |
| EAU, otras (EAU-O) | EAU, no Herlitz, generalizada (EAU-nH gen)*** | |
| EAU, no Herlitz, localizada (EAU-nH loc) | ||
| EAU con atresia pilórica (EAU-PA) | ||
| EAU, inversa (EAU-I) | ||
| EAU, inicio tardío (EAU-lo)**** | ||
| Síndrome laringo-onico-cutáneo (LOC) | ||
| Epidermólisis ampollosa distrófica (EAD) | EAD dominante (EADd) | EADd, generalizada (EADd-gen) |
| EADd, acral (EADd-ac) | ||
| EADd, pretibial (EADd-Pt) | ||
| EADd, pruriginosa (EADd-Pr) | ||
| EADd, solo uñas (EADd-na) | ||
| EADd, dermólisis ampollosa del neonato (EADd-BDN) | ||
| EAD recesiva (EADr) | EADr, grave generalizada (EADr-grave gen)***** | |
| EADr, otras generalizadas (EADr-O) | ||
| EADr, inversa (EADr-I) | ||
| EADr, pretibial (EADr-Pt) | ||
| EADr, pruriginosa (EADr-Pr) | ||
| EADr, centripetalis (EADr-Ce) | ||
| EADr, dermólisis ampollosa del neonato (EADr-BDN) | ||
| Síndrome de Kindler | ||
Variantes poco frecuentes cursiva;
A continuación se describen las manifestaciones clínicas características de los principales tipos de EA (simple, de la unión y distrófica), así como los hallazgos únicos de algunos subtipos. Es importante recordar que las descripciones clásicas de los distintos tipos y subtipos de EA establecen un patrón modelo y que, lejos de ser descripciones rígidas, constituyen guías diagnósticas para el médico. De igual manera, la descripción clínica de los subtipos menos frecuentes se basa en los hallazgos observados en un número limitado de pacientes, por lo cual es posible que el espectro completo de manifestaciones clínicas sea más diverso. Por último, se hará una descripción breve de las nuevas entidades incluidas en el espectro de la EA por el Tercer Consenso Internacional sobre Diagnóstico y Clasificación de la Epidermólisis Ampollosa. Para una descripción más detallada sobre las manifestaciones clínicas específicas de los diferentes subtipos de EA se recomienda leer este consenso5.
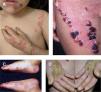
Epidermólisis ampollosa simpleLa mayoría de las EAS son de herencia autosómica dominante, aunque existen subtipos de transmisión recesiva, tales como la EAS autosómica recesiva (EAS-AR), la EAS letal acantolítica, la deficiencia de placofilina, la EAS con distrofia muscular (EAS-MD) y la EAS con atresia pilórica (EAS-PA). Las ampollas aparecen al nacer, se originan tras un evento traumático y frecuentemente aparecen en las palmas y las plantas; son flácidas y al romperse dejan una costra melicérica, sin atrofia o cicatriz. La formación de ampollas ocurre con mayor frecuencia en la infancia y disminuye con la edad. La presentación más común es la EAS localizada (EAS-loc). En caso de presentar ampollas de forma generalizada e hiperqueratosis palmoplantar se debe pensar en la EAS Dowling-Meara (EAS-DM), con compromiso de mucosas y uñas, o en la EAS generalizada no Dowling-Meara (EAS gen-nonDM). La EAS-DM representa la forma más grave de EAS, se manifiesta clásicamente con ampollas herpetiformes y cursa con una mayor mortalidad, principalmente por sepsis. En casos raros se puede presentar EAS con distrofia muscular o con atresia pilórica. La presentación con pigmentación moteada (EAS-MP) es rara5,22–26 (fig. 2).

La EAS-loc es de herencia autosómica dominante debido a un defecto en las queratinas 5 y 14. Clásicamente se limita a las manos y a los pies, se manifiesta por ampollas de base eritematosa originadas por fricción y exacerbadas por la sudoración y el calor excesivos. Con frecuencia está infradiagnosticada, ya que las manifestaciones clínicas no son lo suficientemente graves como para causar preocupación en quienes la padecen5,27.
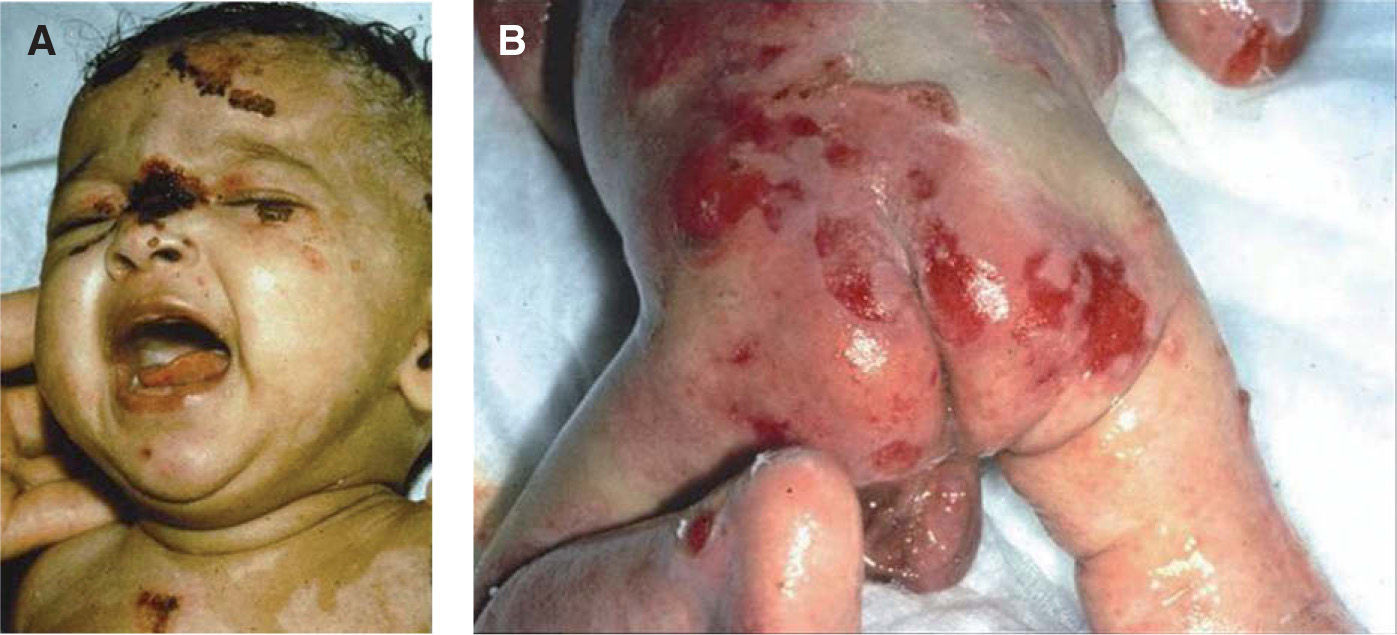
Jonkman et al describieron una nueva forma de EAS llamada letal acantolítica. Esta se debe a una mutación autosómica recesiva en el gen de la desmoplaquina, una proteína de adhesión en las células epiteliales y musculares. La afección es generalizada y las manifestaciones clínicas se observan en el postparto inmediato, caracterizándose por fragilidad cutánea, alopecia universal, onicomadesis y presencia de dientes neonatales. La piel es muy frágil, siendo positivo el signo de Nikolsky. No aparecen ampollas o vesículas, sino erosiones. Se observa regeneración epitelial; sin embargo, esta se desprende rápidamente. Se constatan erosiones conjuntivales y de la cavidad oral, además de alteraciones genitourinarias como pseudofimosis y afección del glande. También pueden existir alteraciones gastrointestinales5,28 (fig. 3).
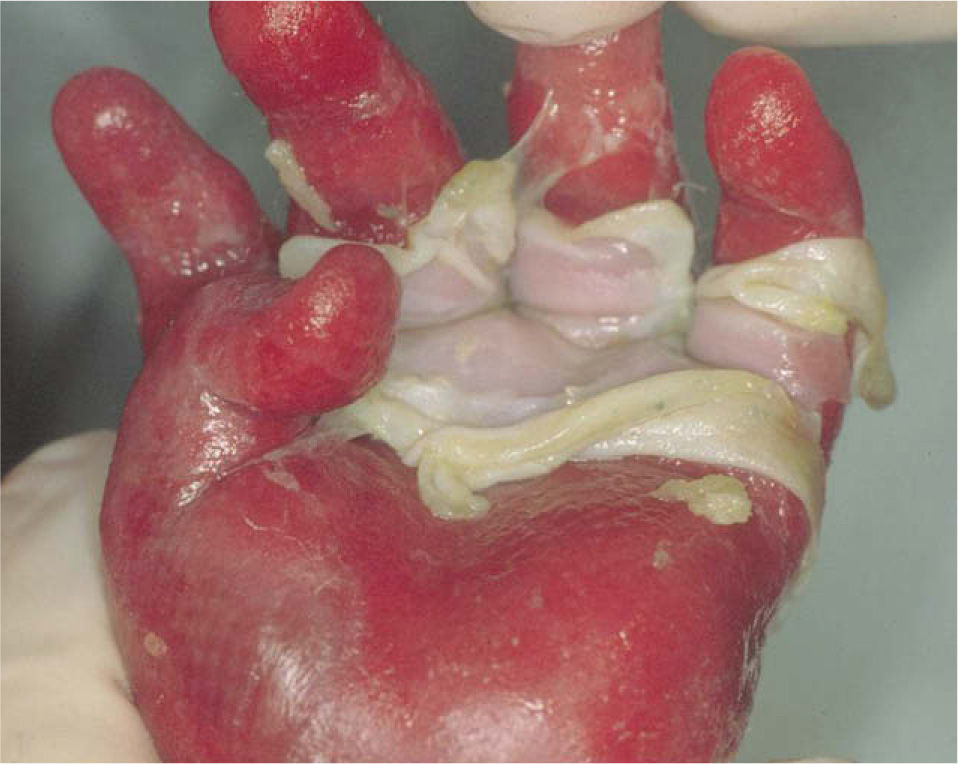
La deficiencia de placofilina o síndrome de McGrath resulta de una mutación del gen PKP1, que trae como consecuencia alteraciones en la placofilina 1, componente estructural de los desmosomas. También se conoce como síndrome de displasia ectodérmica/fragilidad cutánea. Se observa al nacimiento, presentando fragilidad cutánea, formación de ampollas y eritrodermia de manera generalizada. Se acompaña de hipotricosis, distrofia ungueal y queratodermia palmoplantar. Puede existir estreñimiento, estenosis esofágica, blefaritis, fisuras periorales y linguales, así como un moderado retraso del crecimiento. Su patrón de herencia es autosómico recesivo5,29.
La EAS circinada migratoria, causada por una mutación en el gen KRT5 de la queratina, se presenta al nacimiento y es de transmisión autosómica dominante. Se observa eritema migratorio principalmente en las extremidades (manos, pies, piernas y muslos), acompañado de vesículas o ampollas que no dejan cicatriz, pero sí lesiones hiperpigmentadas de color pardo o hipopigmentación5,30.
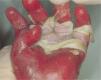
Epidermólisis ampollosa de la uniónLa separación en la EAU ocurre en la lámina lúcida, en la unión dermo-epidérmica. De todos los tipos de EA este es el menos frecuente (fig. 4). La transmisión de este tipo de EA es autosómica recesiva y la distribución de las lesiones puede ser localizada o generalizada. Como regla general, todos los subtipos se caracterizan por presentar ampollas, erosiones cutáneas, distrofia cutánea, hipoplasia del esmalte dental y caries. Al desaparecer, las ampollas dejan cicatrices atróficas. A excepción de la EAU de inicio tardío (EAU-lo), todas se manifiestan en el nacimiento. La afección de las mucosas, sobre todo oral, gastrointestinal, ocular y respiratoria, se presenta fundamentalmente en la EAU tipo Herlitz (EAU-H) y la EAU no Herlitz generalizada (EAU-nH gen). En aquellos subtipos en los cuales están afectadas las mucosas gastrointestinal y respiratoria se encuentra anemia, retraso del crecimiento y alteraciones respiratorias, que se manifiestan inicialmente por estridor o ronquera, pudiendo terminar en fallo respiratorio. La primera causa de muerte en pacientes con EAU es por sepsis, seguida por fallo respiratorio. La mayoría de los decesos se dan en pacientes menores de un año de edad. En el subtipo EAU-PA existe atresia del píloro y malformaciones genitourinarias y puede haber aplasia cutis asociada5,22–24.
La forma más común y grave de EAU es la EAU-H, que se caracteriza por presentar ampollas de distribución generalizada, tejido de granulación hiperplásico en las regiones periorales, perinasales, ungueales o en sitios donde se localizan las ampollas. Afecta a la mucosa de la laringe, bronquios, esófago, recto, vesícula biliar, vagina, tracto urinario y córnea. Se observa hipoplasia del esmalte dental, y por lo tanto, una mayor prevalencia de caries. Puede presentar pseudosindactilia, pero no es muy común. Los pacientes con este subtipo de EAU generalmente no sobreviven a la infancia31,32.
El síndrome LOC se caracteriza por presentar tejido de granulación crónico en la laringe, distrofia ungueal y erosiones cutáneas. Su patrón de herencia es autosómico recesivo y está causado por una alteración en el gen LAMA3A que codifica la proteína laminina alfa 3 A. Las manifestaciones clínicas aparecen al nacimiento, presentando ampollas y úlceras cutáneas distribuidas principalmente en la cara y el cuello, hipoplasia del esmalte dental, simbléfaron y ceguera como consecuencia del tejido de granulación conjuntival. Puede existir anemia secundaria al sangrado de las erosiones, así como alteraciones del tracto respiratorio5,33.
Epidermólisis ampollosa distróficaEstá causada por mutaciones en el colágeno VII que forma las fibrillas de anclaje de la membrana basal epidérmica. Las ampollas pueden ser localizadas o generalizadas, dependiendo del subtipo, y dejan cicatrices distróficas una vez que desaparecen. La transmisión es autosómica dominante o recesiva, siendo las formas recesivas por lo general más graves pero, afortunadamente, menos comunes. Es frecuente la aparición de quistes de milio y distrofia ungueal. El espectro clínico de las formas dominantes va desde la afección localizada hasta la generalizada. En el caso de la EAD dominante pretibial (EADd-Pt), las ampollas aparecen en el área pretibial, los pies, en menor medida en las manos, y puede haber lesiones que semejen un liquen plano. En la EAD dominante acral (EADd-ac) las ampollas sólo aparecen en las manos y los pies. En la EAD dominante generalizada (EADd-gen) se pueden ver pápulas hipopigmentadas en el tronco, llamadas lesiones albopapuloides, las cuales se describieron como clásicas de los antiguos subtipos Cockayne-Touraine o Pasini. Una característica de las formas generalizadas es que conforme avanza la edad, las ampollas tienden a ser más localizadas. La EADd pruriginosa (EADd-Pr) se caracteriza por la presencia de prurito, y en la EADd sólo uñas (EADd-na) únicamente se ven afectadas estas, incluso sin presentarse ampollas. En las formas dominantes, la sintomatología puede estar presente desde el nacimiento, pero también puede manifestarse a partir de la infancia. Las formas recesivas también pueden tener desde espectros leves y localizados (EADr-Pt, EADr-I, EADr-Ce) hasta graves y generalizados (EADr-grave gen, EADr-O). La forma recesiva inversa (EADr-I) afecta de forma localizada, presentándose ampollas en las regiones intertriginosa, lumbosacra, axial y acral, y tiene un riesgo alto de producir estenosis del conducto auditivo externo. La forma recesiva centrípeta (EADr-Ce) afecta a los dedos y a la región pretibial.
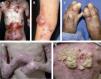
Con mucho, el subtipo más grave es la EADr-grave gen, antes llamada de Hallopeau-Siemens, que se caracteriza por presentar extrema fragilidad de la piel y formación generalizada de ampollas, cicatrización distrófica extensa, pseudosindactilia, distrofia ungueal y dentaria, caries y microstomía. Se encuentran afectadas todas las mucosas, causando estenosis esofágica, anal y uretral, úlceras corneales y fimosis. Se observa anemia y retraso del crecimiento. Los pacientes con EADr-grave gen tienen un riesgo elevado de desarrollar carcinoma de células escamosas de la piel (fig. 5). Otras complicaciones que pueden encontrarse son glomerulonefritis, amiloidosis renal, nefropatía por IgA, insuficiencia renal crónica, cardiomiopatía y osteoporosis. Aparece desde el nacimiento5,22,24,34,35.
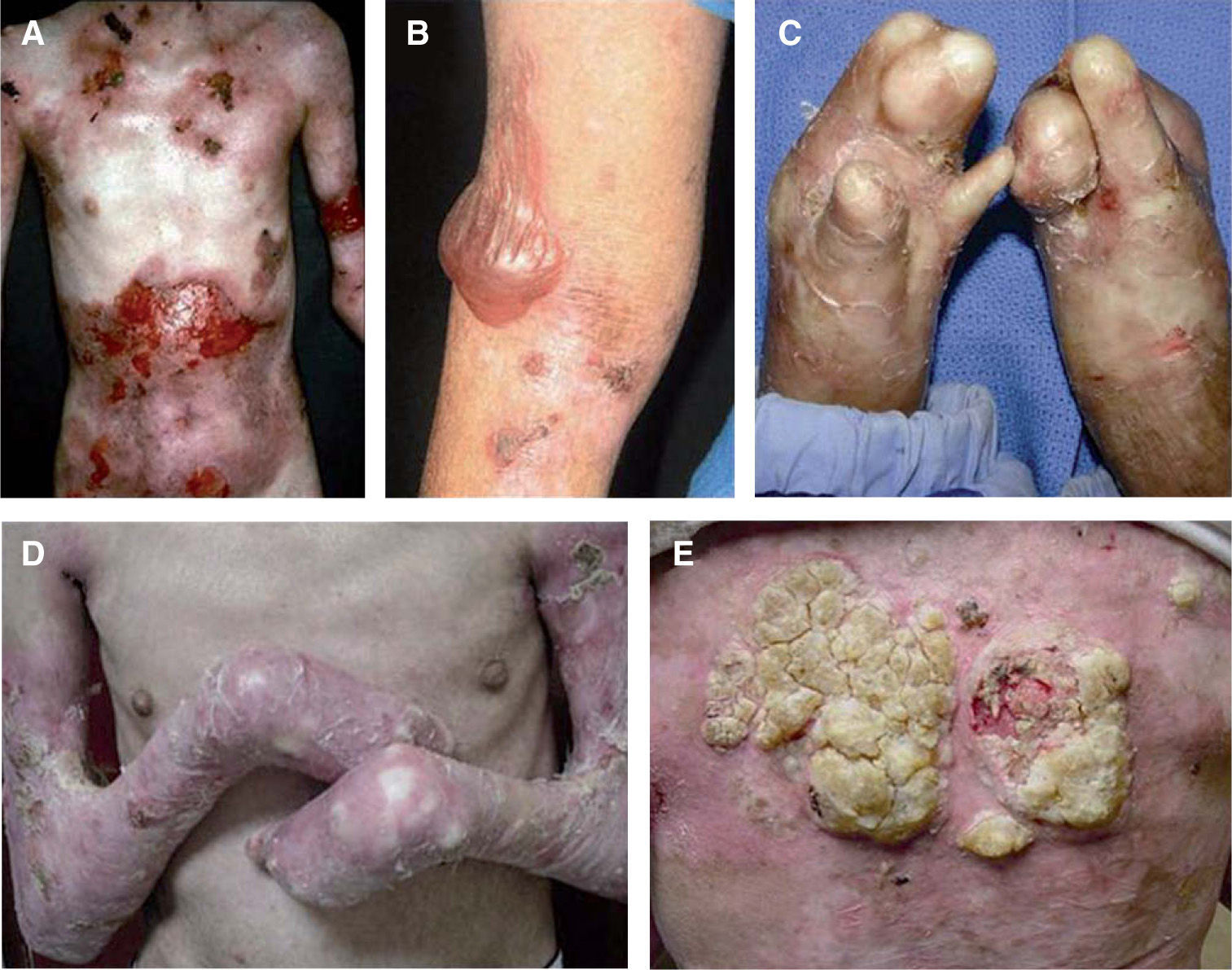
Aspecto clínico de pacientes con epidermólisis ampollosa distrófica (EADr).
A. Grandes áreas denudadas de piel en el tórax.
B. Pueden aparecer ampollas espontáneas en cualquier sitio, sin embargo, son más comunes en lugares de roce.
C. El traumatismo crónico de las manos provoca la formación de cicatrices y contracturas, la aparición de pseudosindactilia, contracturas en flexión y guante de queratina en las manos de un paciente con EADr. D. Contracturas extensas en las articulaciones de codos, manos y rodillas que limitan la movilidad de los pacientes. E. Carcinoma espinocelular agresivo en la espalda de un paciente joven de 24 años de edad.
Es un trastorno autosómico recesivo en el cual hay una pérdida de la proteína kindlina 1 por mutación del gen KIND1. La kindlina tiene como función estabilizar la unión dermo-epidérmica al unir los microfilamentos de actina del citoesqueleto con la matriz extracelular. El síndrome de Kindler se caracteriza por la formación de ampollas de distribución acral y fotosensibilidad en el periodo neonatal y en la infancia, con poiquilodermia en años posteriores. La formación de ampollas puede ser espontánea o traumática, y por lo general se resuelve con la edad. Puede existir atrofia cutánea generalizada, sin embargo, es más común en el dorso de las manos y los pies, en las rodillas y en los codos (fig. 6). Entre otros hallazgos se encuentran: queratodermia palmoplantar, distrofia ungueal, alopecia, estenosis esofágica, anal, vaginal o uretral, fimosis, sindactilia y pérdida de los dermatoglifos9,12,26,36.
Diagnóstico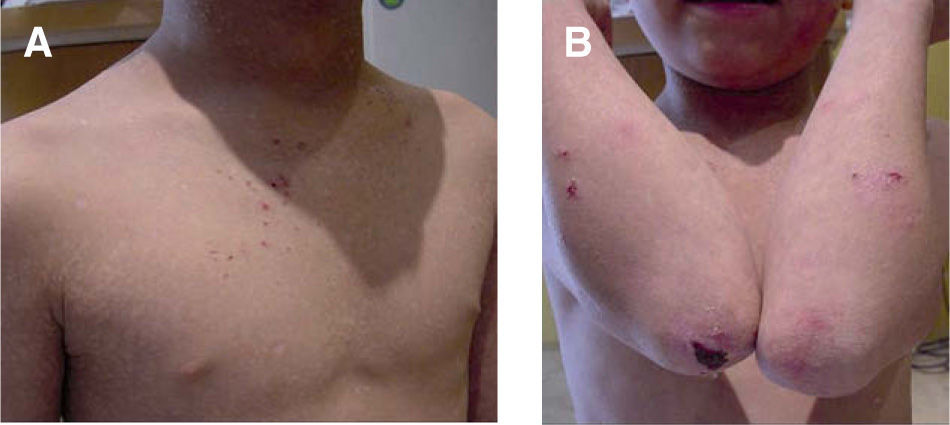
El diagnóstico de EA se sospecha en individuos con piel frágil, y se manifiesta por la formación de ampollas ante traumatismos menores37. Los diagnósticos diferenciales de esta enfermedad incluyen pénfigo vulgar, penfigoide, eccema dishidrótico, dermatosis lineal por IgA, lupus eritematoso ampolloso, picaduras de insectos y ampollas por fricción22. Una vez excluidas otras patologías se procede a confirmar la sospecha de EA mediante inmunofluorescencia o microscopía electrónica para determinar el plano de despegamiento o formación de las ampollas15.
La realización de una historia clínica y exploración física completas constituye el siguiente paso en el diagnóstico, e incluye una evaluación de la distribución de las lesiones y de la presencia de otras características o complicaciones clínicas (tabla 3). En ocasiones se requiere también el análisis genético del paciente y de sus padres para la determinación definitiva del tipo de herencia15,22,23. El objetivo que se persigue es determinar el subtipo de EA que presenta el paciente, lo cual es importante para definir su pronóstico clínico y genético, así como el tratamiento a seguir1,38.
Historia clínica y exploración física en la epidermólisis ampollosa
| Historia clínicaAntecedentes familiares de enfermedades ampollosas ofragilidad de la piel Edad de inicioTamaño, frecuencia y localización de las lesiones ampollosas Posibles factores desencadenantes o agravantes de las lesionesDiagnósticos y tratamientos previosCuantificación del dolor o prurito ocasionado por las lesiones Retraso del crecimiento o desarrollo Afectación de mucosas: síntomas orales, nasofaríngeos, oculares, genitourinarios, gastrointestinales o respiratorios |
| Exploración físicaExploración física completa con énfasis en la piel y lasmucosas oral, genital y conjuntival Evaluar el tamaño, la localización y las características de las ampollasIntentar identificar el sitio de formación de la ampolla Superficial: erosiones y costrasIntraepidérmica: ampollas flácidas que se pueden expandir bajo presiónIntralaminal: ampollas tensas que sanan con atrofia perosin cicatrización Intradérmica: ampollas que sanan con cicatrización y formación de quistes de milio Afección de uñas, cabello o dientes |
Este método diagnóstico se emplea con dos objetivos: determinar el plano de despegamiento e identificar la proteína afectada en un caso específico 19. La biopsia de piel utilizada como muestra se obtiene del borde de una ampolla espontánea o inducida por fricción (por ejemplo, al frotar la piel con el borrador de un lápiz) y se procesa en fresco, con la precaución de tener un medio de transporte adecuado como la solución de Michel5,19,22,39.
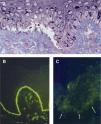
Para determinar el plano de formación de la ampolla se utilizan anticuerpos contra un antígeno del hemidesmosoma (como el antígeno del penfigoide ampolloso BP230) y contra una proteína de la lámina densa (como el colágeno IV). En el caso de la EAS ambos antígenos se localizan en el suelo de la ampolla; en la EAU el antígeno BP230 se localiza en el techo de la ampolla, mientras que el de colágeno IV se localiza en el suelo; en la EAD ambos antígenos se localizan en el techo de la ampolla22. Además, es posible identificar la proteína afectada en un caso específico mediante el análisis de la expresión y distribución de los anticuerpos contra la laminina-332 (anteriormente conocida como laminina-5), el colágeno VII, el colágeno XVII, la plectina, la integrina α6pβ y la queratina 14 (fig. 7).
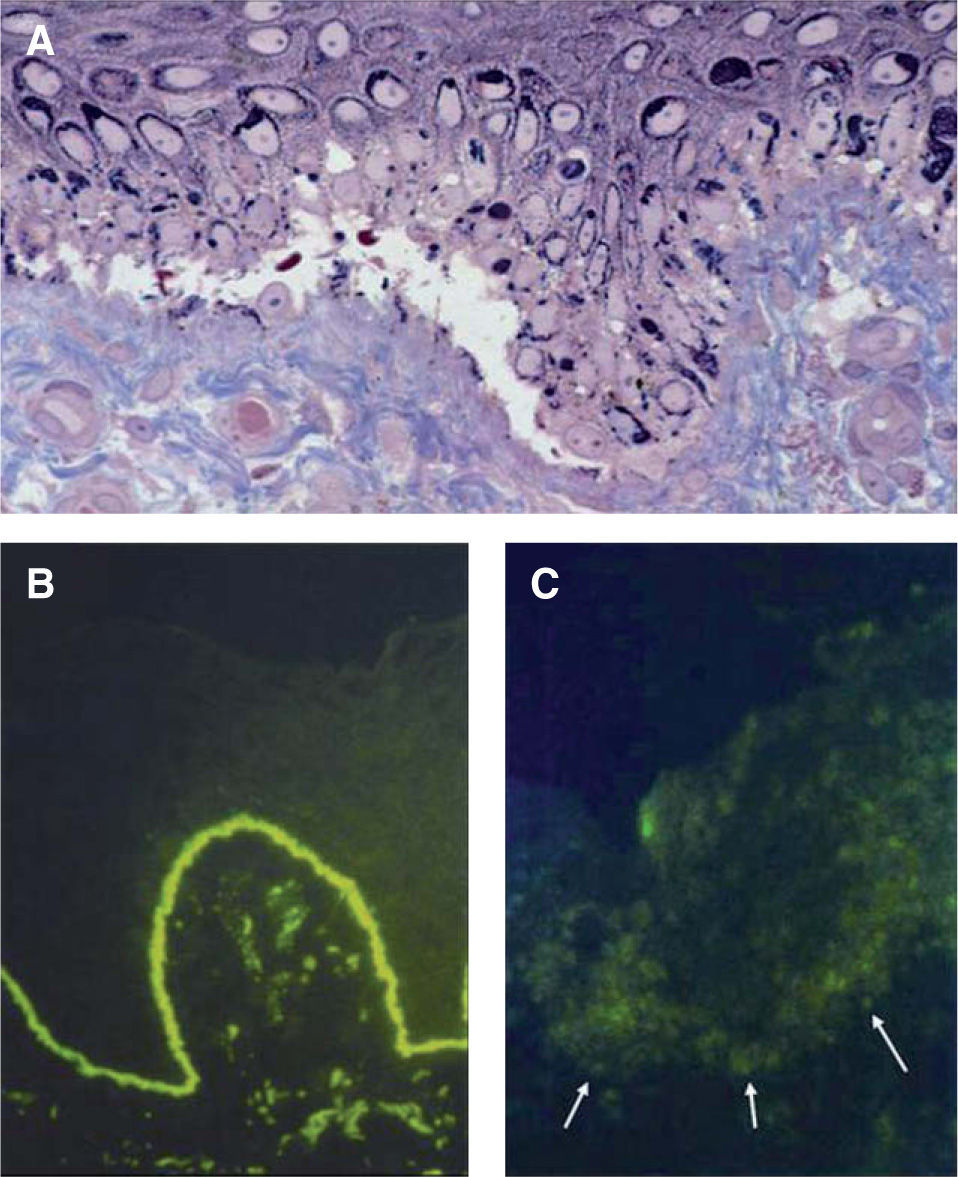
Hallazgos histológicos de las epidermólisis ampollosas en la piel. A. Corte semifino para microscopía electrónica teñido con azul de metileno y Azure II (60x). En la epidermólisis ampollosa simple existe una ampolla dentro de la célula basal epidérmica, por encima de la unión dermo-epidérmica. B. Mapeo por inmunofluorescencia en piel normal (40x) que muestra la presencia de colágeno tipo VII. C. Piel de paciente con epidermólisis ampollosa distrófica recesiva grave, en la que no se observan rastros de colágeno tipo VII por medio del autoanticuerpo contra dicho componente de las fibrillas de anclaje (40x).
A diferencia del estudio de una biopsia de piel por microscopía electrónica, el mapeo por inmunofluorescencia es un método relativamente sencillo, rápido y barato. Como ventaja adicional, el medio de transporte preserva adecuadamente una biopsia de piel durante varias semanas a temperatura ambiente. Por estos motivos se recomienda utilizar el mapeo por inmunofluorescencia como el estudio de laboratorio primario para confirmar el diagnóstico de EA5. Sin embargo, al ser una técnica de análisis semicuantitativo, puede ser imposible diagnosticar la enfermedad en individuos con una producción deficiente (pero no nula) de proteínas de adhesión, por lo que es necesario recurrir a la microscopía electrónica19.
Microscopía electrónicaLa microscopía electrónica es el método de referencia en la clasificación de la EA, dado que puede determinar tanto el nivel del plano de despegamiento como las alteraciones ultraestructurales de las proteínas afectadas (filamentos de queratina, desmosomas, hemidesmosomas, fibrillas de anclaje y filamentos de anclaje), cuyo número o apariencia se ve alterado en los diferentes subtipos de EA5,19. Sin embargo, en la actualidad existen muy pocos laboratorios en el mundo con la experiencia necesaria para realizar un diagnóstico fiable de EA por microscopía electrónica. Debido a esto, se prevé que el uso de esta técnica diagnóstica disminuirá con el tiempo, aunque su aplicación en la investigación es fundamental5.
Análisis de mutacionesEl análisis de mutaciones es el método ideal para determinar el patrón de herencia y la mutación específica causante de la EA en cada caso particular. Esta técnica constituye la base del diagnóstico prenatal y es una excelente herramienta de investigación. Asimismo, el desarrollo de terapias génicas en un futuro se basará en la determinación de las mutaciones específicas presentes en cada subtipo. Sin embargo, en este momento el análisis de mutaciones no se considera como una herramienta diagnóstica de primera línea en la EA5,22.
Diagnóstico prenatalHasta la fecha no existe un tratamiento efectivo para la EA, de manera que el diagnóstico prenatal y el consejo genético son de gran importancia para aquellas parejas con riesgo de transmitir esta enfermedad. En la década de los ochenta se hizo posible el diagnóstico prenatal de la EA mediante la obtención de una muestra de piel fetal por fetoscopia. Actualmente, la toma de una biopsia de piel se puede guiar por ultrasonido y la muestra se analiza por inmunofluorescencia y microscopía electrónica. Este procedimiento se puede realizar a partir de las 15 semanas de gestación, una vez que la piel fetal se ha desarrollado lo suficiente para su análisis.
Los avances en el estudio de la patología molecular de la EA han permitido la introducción de técnicas de diagnóstico prenatal basadas en el análisis del ácido desoxirribonucleico (ADN) fetal a partir de células del líquido amniótico o muestras de vellosidades coriónicas. Tanto la amniocentesis como la biopsia de vellosidades coriónicas conllevan menos riesgo de aborto que la fetoscopia y permiten un diagnóstico más temprano, inclusive a partir de la décima semana de gestación. En ambos casos es necesaria la realización previa del análisis de mutaciones, tanto de los padres como del hijo afectado, y la búsqueda de mutaciones debe realizarse de manera específica para cada familia con riesgo para la EA.
Tanto el análisis de piel fetal como el análisis de ADN fetal tienen como desventaja que presentan como única solución la terminación del embarazo. Una alternativa es la fertilización in vitro, que permite el análisis genético del embrión antes de su transferencia al útero y, por lo tanto, previo al comienzo del embarazo. Es posible realizar un diagnóstico de preimplantación en un embrión en la etapa de 8 células, del cual se extrae una célula para su análisis de mutaciones. Si el análisis revela un genotipo normal, el blastocisto original se implanta en el útero; en caso contrario es descartado. Aunque esta técnica ha sido utilizada en otras enfermedades de fragilidad cutánea, aún no se ha aplicado de manera exitosa en la EA.
Actualmente se encuentran en vías de desarrollo otros métodos diagnósticos menos invasivos, incluyendo el uso de ecografía tridimensional y el análisis del ADN fetal a partir de una muestra de sangre materna19,22,34,40,41.
TratamientoPor el momento no existe una cura definitiva para la EA. El tratamiento de esta enfermedad es sintomático y de soporte, enfocado a prevenir el desarrollo de lesiones y complicaciones. La complejidad del tratamiento dependerá de la gravedad de las lesiones que presente el paciente. Se requiere un equipo multidisciplinario para el manejo óptimo de estos pacientes que comprenda las disciplinas de Dermatología, Cirugía, Nutrición, Odontología, Fisioterapia, Enfermería, Psicología, Medicina del Dolor y Genética (tabla 4). El plan de tratamiento se debe individualizar en cada caso, y una óptima comunicación entre los miembros del equipo es vital para la obtención de buenos resultados.
Manejo y tratamiento de la epidermólisis ampollosa y de sus complicaciones
| Oftalmología | Preventivo | Examen oftalmológico inicial y, si está indicado, repetir anualmente |
| Lágrimas artificiales con metil celulosa como prevención de abrasiones corneales | ||
| Uso de lentes de contacto suaves para prevenir erosiones corneales | ||
| Tratamiento | Exploración urgente en caso de ojo rojo, fotofobia o sensación de cuerpo extraño | |
| Uso de lubricantes oculares libres de lanolina para xeroftalmía | ||
| Antibióticos tópicos y parches oculares en caso de erosiones corneales | ||
| Lubricantes oculares y cámaras de humedad para el tratamiento de la blefaritis crónica | ||
| Odontología | Preventivo | Intervención dental temprana y agresiva |
| Limpiezas anuales/bianuales con aplicación de flúor | ||
| Uso de cepillo de dientes de cabeza pequeña y cerdas suaves, previamente remojado en agua caliente | ||
| Ejercicios para la prevención de microstomía y anquiloglosia | ||
| Tratamiento | Enjuagues con sucralfato disminuyen el dolor de las lesiones orales y previenen la formación de ampollas | |
| Restauración dental bajo anestesia general en caso necesario | ||
| Nutrición | Preventivo | Uso de biberones para labio y paladar hendido en bebés |
| Vigilancia de los parámetros de crecimiento, especialmente las curvas de IMC/crecimiento | ||
| Involucrar a un nutricionista clínico desde el inicio | ||
| Seguimiento cada 6 a 12 meses con los siguientes exámenes de laboratorio: biometría hemática completa, urea, electrolitos, pruebas de función hepática, velocidad de sedimentación globular, proteína C reactiva, hierro, ferritina, calcio, fosfato, vitamina D, zinc, selenio | ||
| Tratamiento | Comidas frecuentes, con alto contenido calórico, de consistencia blanda, no ácidas y no calientes | |
| Requerimientos proteicos y calóricos del doble de lo recomendado para la edad o de acuerdo a la fórmula de Birge | ||
| Multivitamínicos 1 o 2 veces la dosis recomendada | ||
| Suplementos de calcio y vitamina D para la osteopenia-osteoporosis | ||
| Hierro oral (¡cuidado! puede causar molestias gastrointestinales y estreñimiento), hierro intravenoso más eritropoyetina, transfusiones sanguíneas para la anemia | ||
| Quirúrgico | Manejo periquirúrgico | ¡Nunca poner adhesivos directamente sobre la piel! |
| Colocar al paciente sobre una superficie amortiguadora de puntos de presión | ||
| Gastrostomía | Colocación abierta o percutánea, con técnica endoscópica o no endoscópica. Se reemplaza con botones | |
| Pseudosindactilia | Liberación quirúrgica de pseudosindactilia y contracturas en flexión desde el momento en que exista un compromiso serio de la función | |
| El paciente debe cumplir un régimen posquirúrgico estricto que incluya rehabilitación, uso de guantes y férulas rígidas | ||
| La función recuperable no es óptima | ||
| Es inevitable la recurrencia en 1 a 3 años | ||
| Carcinoma espinocelular | Escisión de lesiones malignas | |
| Heridas | Ampollas | Puncionar su base con aguja estéril y aplicar presión sobre la ampolla con una gasa para exprimir el líquido y prevenir la extensión de las ampollas, sin retirar el techo de la ampolla |
| Erosiones y úlceras | Curaciones diarias, aplicando antibiótico tópico seguido de un vendaje no adherente, como gasa con vaselina, cubierto con gasa seca | |
| Humedecer o mojar los vendajes antes de cambiarlos | ||
| Infección | Prevención | Cambios diarios de vendajes |
| Uso de agentes tópicos: antibióticos, ácido acético, cloro, compuestos de plata, peróxido de hidrógeno, miel | ||
| Rotación mensual de antibióticos tópicos para prevenir resistencias | ||
| Tratamiento | Antibióticos sistémicos | |
| Cultivo de heridas y antibiograma para guiar el tratamiento | ||
| Gastroenterología | En caso de estreñimiento: administrar laxantes y ablandadores de heces y aplicar petrolato en el área perianal para evitar fisuras | |
| En caso de estenosis esofágica: dilatación esofágica con globo y gastrostomía si esta no tiene éxito. También es posible hacer dilataciones anterógradas o retrógradas a través de la gastrostomía | ||
| Rehabilitación | Para la prevención de contracturas articulares, microstomía y anquiloglosia | |
| Uso de guantes elásticos para prevenir la pseudosindactilia | ||
| Psicología | Apoyo psicológico para pacientes y familiares con atención a la aceptación de la enfermedad, evitar sobreprotección y aislamiento de los pacientes | |
| Manejo de crisis como depresión e intentos de suicidio | ||
| Anestesia | Manejo del dolor | Analgésicos orales para el dolor causado por ampollas agudas |
| Quirúrgica | Se prefiere siempre que sea posible la anestesia local o regional | |
| En caso de anestesia general con intubación endotraqueal, tener mucho cuidado con la mucosa oral y faríngea durante la intubación y extubación |
IMC: índice de masa corporal.
Mantener la salud oral promueve una mucosa saludable y mejora la masticación, la deglución, la nutrición, la respiración y el habla. Una rutina diaria de rehabilitación evita la formación de contracturas articulares. El apoyo psicológico a los padres y familiares es básico, y se debe buscar evitar la sobreprotección del paciente. La EA no representa una contraindicación para la aplicación de cualquier inmunización.
La educación de los pacientes con EA y de sus familiares es la piedra angular en su tratamiento. Se recomienda sugerir el acceso a grupos de apoyo como la Dystrophic Epidermolysis Bullosa Research Association (DebRA) para la obtención de información adecuada en español sobre sus cuidados, así como aportar una carta que explique la naturaleza no contagiosa de la enfermedad para los fines que al paciente le convengan42–59.
Dystrophic Epidermolysis Bullosa Research AssociationMundialmente existen organizaciones voluntarias y sin fines de lucro que se dedican a brindar apoyo e información a las personas con EA, a sus familias y al equipo médico involucrado en su cuidado. La organización DebRA, con grupos en más de 32 países, ofrece excelentes manuales informativos sobre los cuidados que requiere un paciente con EA, y cuenta con los contactos de médicos con experiencia en el cuidado de pacientes con esta enfermedad. Muchas de estas organizaciones también contribuyen a la investigación básica y clínica sobre la EA4 (tabla 5).
Algunas asociaciones de la Dystrophic Epidermolysis Bullosa Research Association en el mundo
| DebRA InternacionalDebRA Inglaterra | www.debra-international.orgwww.debra.org.uk |
| Información en españolDebRA MéxicoDebRA AméricaDebRA ChileDebRA EspañaDebRA Costa Rica | www.debra.org.mxwww.deba.orgwww.debrachile.clwww.aebe-debra.orgwww.debracr.org |
En la actualidad, la carrera por encontrar la cura a esta genodermatosis ha desencadenado un gran interés en todo el mundo. En un tratamiento ideal para las EA el objetivo final es la corrección de la deficiencia o ausencia de proteínas de anclaje específicas en la unión dermo-epidérmica. Con este fin en mente se encuentran en estudio diversas terapias proteicas, celulares y genéticas9. Las principales investigaciones en el tema están en continua evolución. Por ejemplo, la terapia génica con cremas o con injertos de células epidérmicas ha sido sustituida por el uso de inyecciones intradérmicas de fibroblastos y la aplicación de células madre del propio paciente. Ultimamente se está investigando en lo que parece ser una luz en el camino de estos pacientes: el trasplante de médula ósea de hermanos sanos inmunocompatibles.
Terapia proteicaConsiste en aplicar directamente la proteína faltante, que se obtiene por métodos recombinantes. Se ha observado que la aplicación tópica de colágeno VII humano en ratones con el gen Col7a1 inactivado promueve una curación más rápida de las heridas. La administración intradérmica o intravenosa de colágeno VII humano en ratones con el gen Col7a1 inactivado genera niveles elevados de esta proteína en la membrana basal y fibrillas de anclaje, aumenta la tasa de curación de las heridas e incluso prolonga la supervivencia de dichos ratones.
Se han obtenido resultados similares mediante el uso de mini-colágeno, producido al remover una porción del dominio colagenoso del colágeno VII, lo cual hace que la proteína sea menos susceptible a la digestión proteica. Tras la inyección intraepidérmica en ratones con el gen Col7a1 inactivado se detectó expresión e incorporación del colágeno VII en las fibrillas de anclaje, una reducción en el fenotipo y mayores tasas de supervivencia20,58.
La investigación actual se centra sobre todo en la EAD y en la restitución del colágeno VII faltante; sin embargo, también se han hecho experimentos que sustituyen la laminina 332, la cual es deficiente o está ausente en la EAU9. Por el momento sólo se experimenta con modelos murinos.
Terapia celularConsiste en el uso de células, aplicadas de forma local o sistémica, con el fin de que estas produzcan las proteínas de la unión deficientes o que se diferencien de otras líneas celulares que puedan realizar dicha función. En este momento las células madre y los fibroblastos son las opciones más prometedoras.
Se ha observado en ratones sometidos a un trasplante de médula ósea cómo las células madre hematopoyéticas migran hacia la piel y se diferencian en células madre epidérmicas, las cuales a su vez producen queratinocitos. Además de explicar el posible origen de las enormes cantidades de células madre necesarias para mantener el proceso de curación a lo largo de la vida en los pacientes con EA, estas observaciones señalan el trasplante de médula ósea como una herramienta terapéutica para la EA3,58. Actualmente, la Universidad de Minnesota se encuentra realizando un protocolo de investigación que evalúa el uso de quimioterapia seguido de trasplante de células madre hematopoyéticas de donante alogénico para el tratamiento de pacientes con EAD; los resultados preliminares reportados son alentadores60,61.
El uso de fibroblastos en la terapia celular es atractivo debido a que su cultivo y expansión son simples, son más consistentes y se pueden congelar y ser manipulados y aplicados de forma más simple que los queratinocitos9,20. Además, se ha reportado que los fibroblastos dérmicos por sí solos son capaces de producir colágeno VII en la unión dermo-epidérmica62.
Wong et al administraron fibroblastos alogénicos por vía intradérmica en pacientes con EADr, observando un aumento en la producción de colágeno VII de morfología anormal propio del paciente, así como la formación de fibrillas de anclaje rudimentarias que proporcionaron un incremento en la adhesión de la unión dermo-epidérmica. Se piensa que por medio de una señal paracrina los fibroblastos autólogos son capaces de incrementar la síntesis de colágeno VII mutante, el cual, en cantidades suficientes, puede reducir la formación de ampollas63.
El uso de células madre mesenquimales también se encuentra bajo estudio y se ha comunicado que su aplicación en pacientes con EADr tuvo como resultado una mejoría en la curación de las heridas y en la expresión de colágeno VII en la unión dermo-epidérmica58.
Terapia génicaConsiste en la corrección directa del genotipo enfermo. Esto se realiza al transferir a la célula uno o más genes externos, los cuales pueden o no integrarse al genoma, y que tienen como objetivo la adición, el reemplazo o la supresión de funciones que, en su estado«natural», causan el fenotipo de la EA. Para que la terapia génica sea una opción realista se debe prestar atención a los siguientes asuntos: a) determinar qué células deben tratarse, b) qué abordaje debe usarse (ex vivo o in vivo) y c) qué vector debe ser utilizado (virales o no virales)64.
En el abordaje in vivo el material genético se introduce directamente a través de la piel, ya sea mediante un vehículo tópico, inyección, electroporación o por medio de biolística (gene gun). La desventaja del abordaje in vivo es que la transferencia de genes tiene baja eficiencia y la síntesis del producto deseado dura muy poco. En contraposición, el abordaje ex vivo implica la realización de una biopsia de piel seguida de su expansión in vitro, la introducción de material genético en las células y el retorno de la piel modificada y expandida al paciente en forma de injertos20,64.
La transferencia de genes puede realizarse por vectores no virales y virales. Los plásmidos y transposones se incluyen entre los vectores no virales, son capaces de transferir segmentos largos de ADN; sin embargo, su mayor desventaja es que tienen una baja eficiencia de transducción y una expresión transitoria3. Los vectores virales son más efectivos, siendo los retrovirus y lentivirus los más prometedores actualmente. Entre las desventajas del uso de vectores virales cabe mencionar que no pueden transferir segmentos largos de ADN, además de ser potencialmente oncogénicos e inmunogénicos9,20,62,65.
Se ha reportado que la transferencia de genes mediante cotransfección con plásmidos codificadores de la integrasa FC31 y del colágeno VII o de la laminina 332 resultó en la expresión de colágeno VII en queratinocitos con EAD y de laminina 332 en queratinocitos con EAU in vitro9.
La transferencia del gen COL7A1 mediante vectores lentivirales en queratinocitos y fibroblastos de genotipo EAD in vitro resultó en una restauración de la estructura de la membrana basal, así como expresión de colágeno VII. La transferencia de genes usando vectores retrovirales ha tenido cierto éxito en modelos animales de EAU con mutaciones LAMB3, ITGB4 y COL17A1, así como en modelos caninos de EAD 3,20. Asimismo, se reporta la transferencia exitosa de un mini-gen recombinante de COL7A1, lográndose la expresión de la proteína in vitro20.
Recientemente, se ha probado el uso de terapia génica como tratamiento para EAU en humanos. Se obtuvieron células madre epidérmicas de un paciente adulto con EAU que tenía una deficiencia de la cadena β3 de la laminina 332. A estas células se les transfirió cDNA del gen LAMB3 por medio de un vector retroviral, las cuales fueron cultivadas para la preparación de injertos de piel. Posteriormente, estos injertos se implantaron al paciente. Mediante el análisis histológico de las biopsias de piel se observaron niveles normales de laminina 332, así como una correcta adhesión de la epidermis, condición que permaneció estable durante un periodo de seguimiento de un año66.
Seguramente los avances de la ciencia y el entusiasmo de los médicos e investigadores que han dedicado gran parte de sus vidas al estudio de las EA darán frutos, aportando nuevos descubrimientos que brinden a los pacientes que padecen esta enfermedad una mejor calidad y esperanza de vida.
Conflicto de intereses
Declaramos no tener ningún conflicto de intereses.
Dres. John McGrath, Marcel Jonkman, Angélica Beirana y licenciado Alvaro de Luna.


