



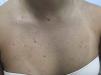
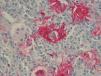
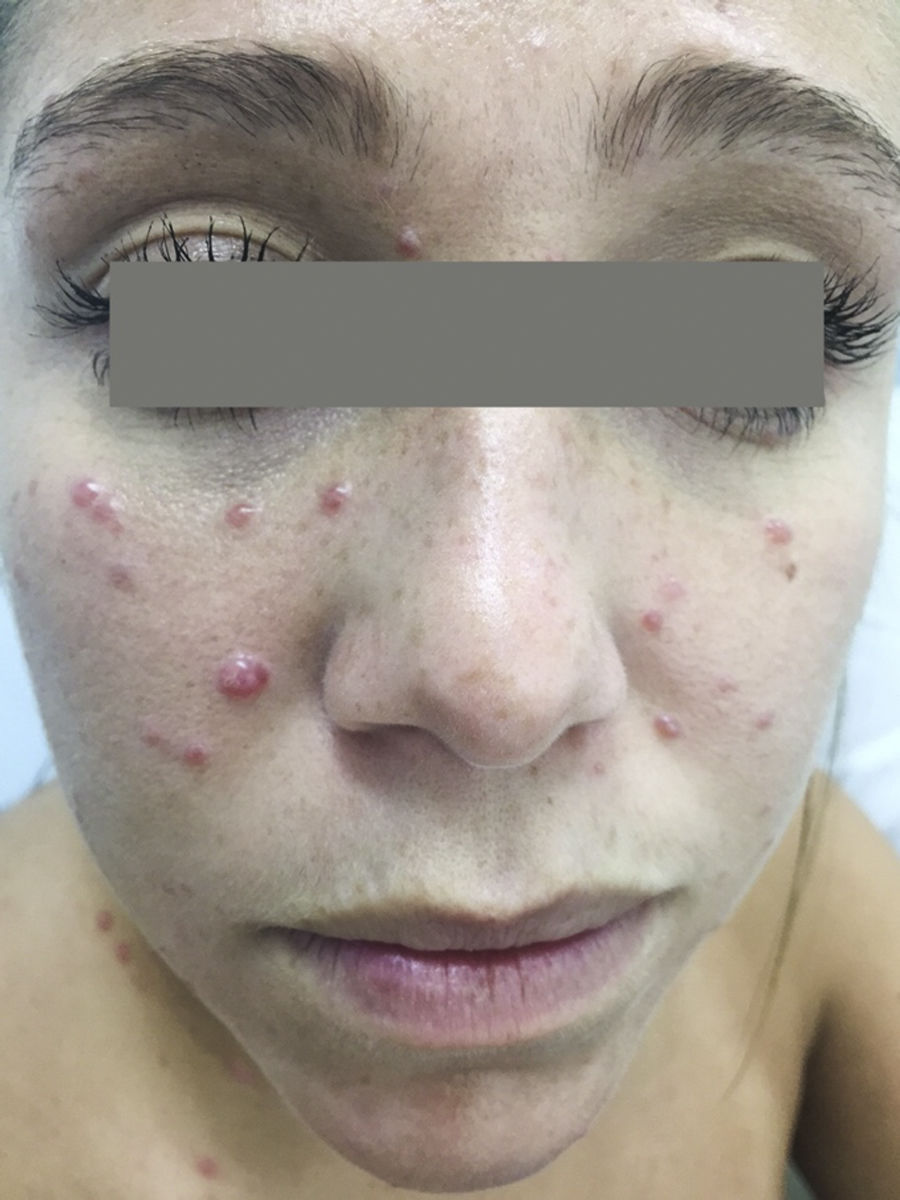
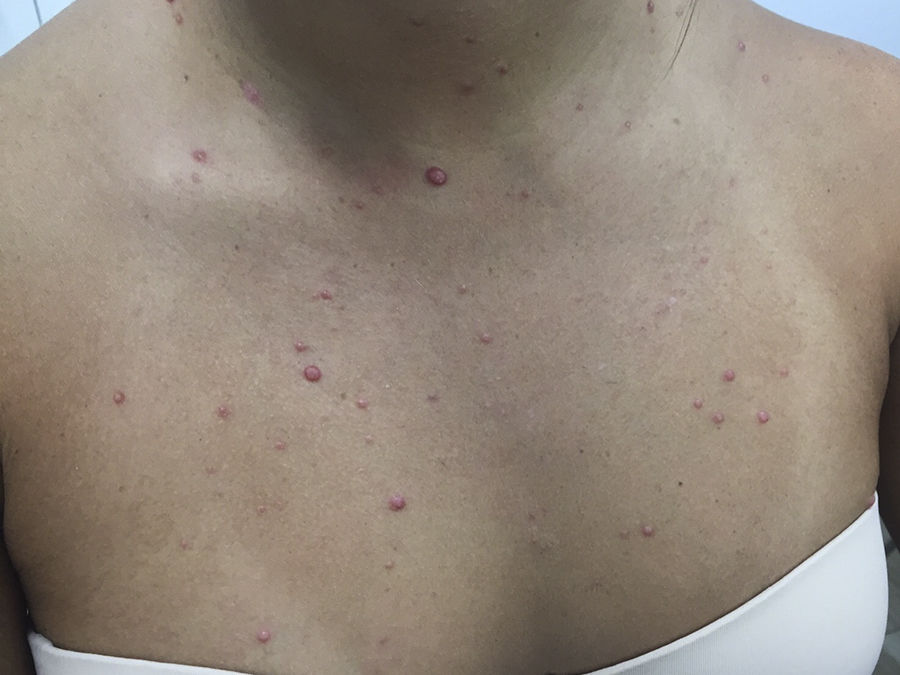
A 21-year-old woman presented with a 3-month history of a diffuse asymptomatic eruption on her face and trunk. She denied fever or any other constitutional symptoms and her review of symptoms was non-contributory. She denied any pertinent past medical or family history. Her only medication included norgestimate/ethinyl estradiol which she had been taking for several years. Physical examination revealed diffuse scattered non-follicular based flesh-colored papules and small nodules. Many lesions demonstrated a central indentation resembling molluscum contagiosum (see Figures 1 & 2). She did not have any cervical, axillary, or inguinal lymphadenopathy. Mucous membrane examination was unremarkable and lacrimal glands did not appear enlarged. Routine histologic examination of her right neck lesion revealed a dense nodular dermal mononuclear cell rich infiltrate showing a significant number of plasma cells and numerous scattered S100 positive multinucleated histiocytes with marked emperipolesis and inconspicuous eosinophils (see Figure 3). Complete blood count with differential, erythrocyte sedimentation rate, lactate dehydrogenase, liver transaminases, alkaline phosphatase, bilirubin, and creatinine were negative or within normal limits. Chest and abdominal magnetic resonance imaging were normal. She was treated with a 60mg oral prednisone taper over six months which resulted in complete resolution of her skin lesions. Follow up at 10 months from onset of disease demonstrated complete remission.
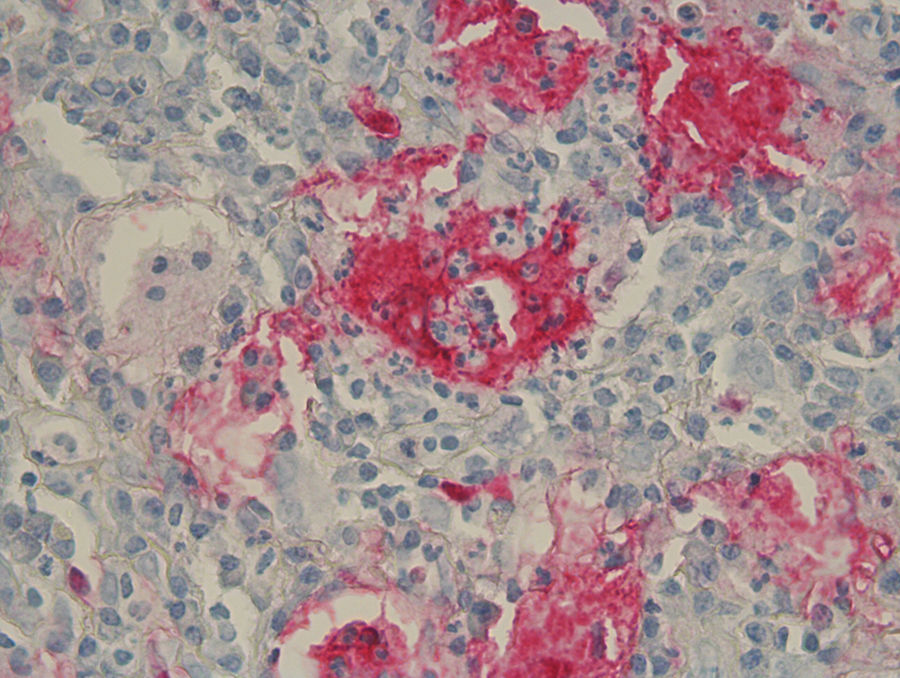
Rosai-Dorfman Disease (RDD) is a relatively rare histiocytic proliferation disorder that was first described in 1969.1 Though RDD classically presents with bilateral massive lymphadenopathy and systemic symptoms, it typically has a benign clinical course and favorable prognosis. In RDD, 43% of patients have involvement of other extranodal sites with skin being the most common site.2 Though skin comprises 10% of extranodal sites involved, approximately 3% are solely cutaneous Rosai-Dorfman Disease (CRDD) without any nodal or other extranodal sites.3 While incidence is reportedly low, CRDD is more prevalent in middle-aged White and Asian women.2 The etiology of RDD is unclear although immunologic,4 viral, and genetic causes including SLC29A3 mutations have been hypothesized.5
The classic presentation of CRDD is a relatively asymptomatic self-involuting nodulo-plaque with surrounding satellite papules.3 However, an evolving wide spectrum of clinical morphologic presentations have been reported. The most common site involved is the face, followed by thigh, and trunk. Recurrence has been reported to occur within 1 to 3.5 years.6 To the best of our knowledge, this will be the first description of RDD mimicking molluscum contagiosum.
The pathognomonic histologic finding of RDD is emperipolesis of intact lymphocytes by S100 and CD68 positive as well as CD1a and langerin negative pale histiocytes characterized also by vesicular nuclei and small nucleoli. Of note, emperipolesis entitles the presence of engulfing of intact hematologic cells, as opposed to hemophagiocytosis which degrades engulfed cells, by histiocytes or megakaryocytes. The so called “Rosai-Dorfman cell” is a type of histiocyte characterized by empiropolesis of only haloed lymphocytes.
These characteristic histiocytes can be abundant or inconspicuously scattered amidst other inflammatory cells.7 Plasma cells are virtually present in all lesions of CRDD. Polymorphonuclear cells, namely eosinophils, are frequently found in skin lesions. Increased vascularity with plump endothelium, with or without fibrosis, are not uncommonly identified namely in long standing lesions.
Emperipolesis in cutaneous disorders has been traditionally related to RDD until recent descriptions of other disorders related to the so-called H syndrome.8 This disorder is characterized by hyperpigmentation, hypertrichosis, hearing loss, heart anomalies, hepatosplenomegaly, hypogonadism, and hallux valgus9 and is caused by a mutation in the SLC29A3 gene. Many authors believe that syndromes associated with mutations in SLC29A3 including familial RDD and pigmented hypertrichosis with insulin-dependent diabetes,5 in addition to H syndrome, fall into the same spectrum of RDD.
Recognition of the pleomorphic genetic and phenotypic presentations of SCL29A3-related diseases is important for diagnosis and for consideration in the histopathologic differential diagnosis of emperipolesis. In addition to SCL29A3-related diseases, IgG4-related diseases have also been shown to have some overlap with RDD as a subset of RDD has been found to contain increased numbers of IgG4-positive plasma cells.4 However, much controversy remains regarding this.
Currently there is no standard guideline for the management of CRDD. The clinical course of CRDD is usually benign and self-limited. Spontaneous resolution varies and ranges from months to several years.2 A wide spectrum of therapeutic interventions including surgical excision, cryotherapy, radiotherapy, lesional and systemic corticosteroids, thalidomide, methotrexate, and even chemotherapy10 have also been reported to be successful for resistant and/or recurrent lesions.
In summary, we first present a case of CRDD with a peculiar eruption resembling molluscum contagiousum thus expanding the spectrum of the cutaneous clinical presentation of the disease and making CRDD another dermatologic masquerader. The nonspecific clinical presentation along with the not infrequently inconspicuous finding of Rosai-Dorfman cells in skin samples and furthermore the typical spontaneous resolution may represent clinico-pathologic characteristics that lead to the tangible possibility of CRDD being underreported.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Conde JM, Kim AY, de Miguel R, Nousari CH. Enfermedad de Rosai-Dorfman cutánea: una nueva presentación clínica. Actas Dermosifiliogr. 2018;109:655–657.


