













La epidermólisis bullosa (EB), enfermedad genética de fragilidad mucocutánea rara y devastadora, es clínica y genéticamente heterogénea. Se caracteriza por la aparición de ampollas inducidas por contacto/fricción o de forma espontánea. La EB se clasifica en 4 tipos: simple, juntural, distrófica y síndrome de Kindler y en 30 subtipos. Esta genodermatosis está causada por defectos en proteínas implicadas en la adhesión dermoepidérmica, con al menos 19 genes caracterizados hasta el momento y más de 1.000 mutaciones identificadas, que explican la complejidad de su diagnóstico. El diagnóstico molecular de la EB es el último paso de un proceso laborioso que se inicia con la recogida de una historia clínica detallada y la toma de una biopsia cutánea, que incluya una zona de despegamiento entre la dermis y la epidermis inducida, en el momento de la recolección. Dicho despegamiento permite establecer el plano de rotura por mapeo antigénico y, en el mejor de los casos, un único gen candidato en el que realizar la búsqueda de las mutaciones patogénicas. Finalizado el diagnóstico molecular, se está en condiciones de ofrecer al paciente un asesoramiento genético adecuado (patrón de herencia, riesgo de recurrencia y opciones de diagnóstico prenatal y preimplantacional) y los consecuentes programas preventivos, así como un pronóstico clínico razonable que facilite su acceso a opciones terapéuticas y de rehabilitación específicas. Por último, el diagnóstico molecular es imprescindible para la participación de los pacientes en ensayos clínicos, de gran importancia en una enfermedad como la EB, que no tiene cura. El objetivo de la presente guía es difundir el procedimiento de diagnóstico de la EB tal y como se está llevando a cabo en nuestro laboratorio y, así, evitar diagnósticos clínicos subóptimos o incompletos. Las recomendaciones recogidas son fruto de nuestra experiencia de más de 10 años de diagnóstico molecular de EB en España.
Epidermolysis bullosa (EB) is a rare genetic disease that causes mucocutaneous fragility. It comprises a clinically and genetically heterogeneous group of disorder characterized by spontaneous or contact/friction–induced blistering. EB is classified into 4 types–simplex, junctional, dystrophic, and Kindler syndrome—and 30 subtypes. The disease is caused by defects in proteins implicated in dermal-epidermal adhesion. At least 19 genes have been characterized and more than 1000 mutations identified, thus rendering diagnosis complex. Molecular diagnosis of EB is the last stage of a laborious process that starts with a detailed clinical history compilation and careful procurement of a skin fresh biopsy that includes an area where the epidermis detaches from the dermis. The detachment area makes it possible to establish the cleavage plane by antigen mapping and, in the best scenario, to identify a single candidate gene to search for pathogenic mutations. The results of the molecular diagnosis enable the physician to provide appropriate genetic counseling (inheritance pattern, risk of recurrence, and options for prenatal and preimplantation diagnosis) and implement subsequent preventive programs, as well as to establish a reasonable clinical prognosis facilitating access to specific therapy and rehabilitation. Lastly, molecular diagnosis is essential for the participation of patients in clinical trials, a critical issue given the current incurable status of EB. The present guidelines aim to disseminate the procedure for diagnosing EB in our laboratory and thus avoid suboptimal or incomplete clinical diagnoses. The recommendations we provide are the result of more than 10 years’ experience in the molecular diagnosis of EB in Spain.
Bajo la denominación de epidermólisis bullosa (EB), comúnmente llamada «piel de mariposa», se encuentra un grupo genética y clínicamente heterogéneo de enfermedades raras, caracterizadas por una extrema fragilidad de la piel y de las mucosas. Este trastorno da lugar a la formación de ampollas y erosiones que se generan por fricción o incluso de forma espontánea1,2. Otras complicaciones asociadas a la EB son la aparición de lesiones hipoplásicas del esmalte dental; estenosis o estrechamiento de las vías respiratorias, gastrointestinales y urogenitales; atresia pilórica; distrofia muscular y cáncer2-4. Por lo tanto, la atención de estos pacientes requiere de un equipo multidisciplinar (para más información consultar la «Guía de atención clínica integral de la epidermólisis bullosa hereditaria» publicada por el Ministerio de Sanidad y Consumo5).
La EB afecta a personas de todos los orígenes étnicos y por igual a ambos géneros. Se estima que en Estados Unidos la incidencia global es de 1/53.000 y la prevalencia 1/125.000. Estimaciones similares se han obtenido en algunos países europeos, incluida España6 (1/166.000; www.orpha.net).
La EB se considera una enfermedad huérfana, ya que no existe en la actualidad una terapia específica y efectiva, y los tratamientos disponibles son únicamente sintomáticos7. La comunidad científica está haciendo un gran esfuerzo por ofrecer soluciones innovadoras basadas en las llamadas terapias avanzadas: terapia celular, génica e ingeniería de tejidos, así como la terapia proteica8-12.
Principales tipos de epidermólisis bullosaSe definen 4 tipos de EB según el plano de fractura cutánea cuando se induce una ampolla: EB simple (EBS), EB juntural (EBJ), EB distrófica (EBD) y síndrome de Kindler (SK). En la EBS la fragilidad se detecta a nivel intraepidérmico (que puede afectar tanto a los queratinocitos basales como suprabasales). En la EBJ la formación de ampollas se produce en la lámina lúcida y en la EBD, por debajo de la lámina densa (en la dermis papilar). En el SK la fractura puede ocurrir a cualquier nivel, excepto en los estratos suprabasales2(fig. 1).
Etiopatogenia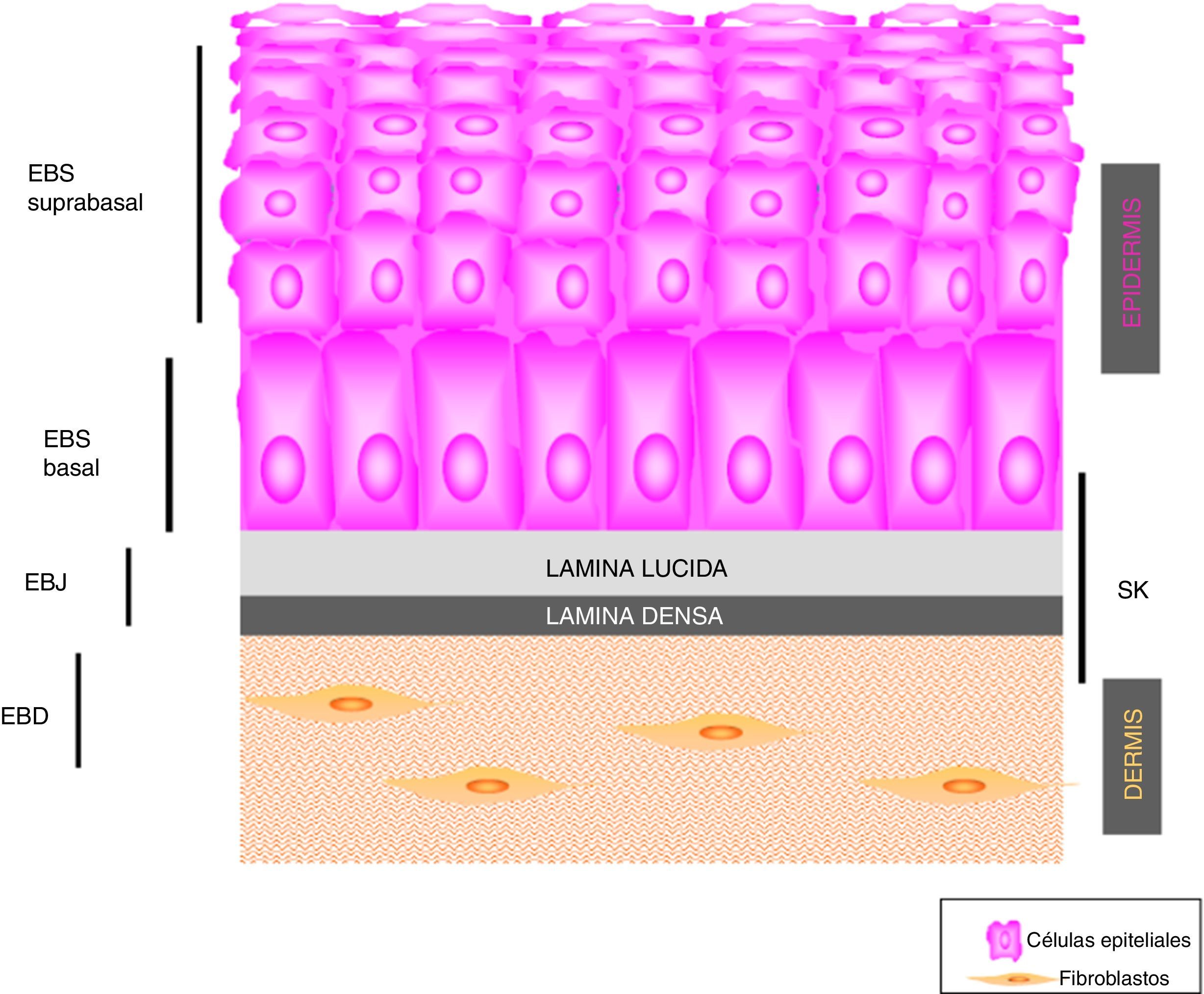
La EB está causada por mutaciones en genes que codifican proteínas responsables de la integridad y la estabilidad mecánica del tegumento13-15. Estas proteínas pueden ser intracelulares, transmembrana o extracelulares y están implicadas en la formación del citoesqueleto, de la unión célula-célula o de las interacciones célula-matriz. La EB presenta un modelo de herencia autosómica que puede ser dominante o recesivo. El gran número de proteínas que forman parte de las estructuras adhesivas de la piel explica la heterogeneidad clínica de la enfermedad. Se han documentado más de 1.000 mutaciones en al menos 19 genes asociados a la patogenia de la EB (tabla 1). Recientemente, se ha descubierto un nuevo gen causante de fragilidad cutánea KHLH 2413. Este gen codifica para la proteína kelch like 24 y causa un nuevo tipo de EBS basal que no está incluido en la clasificación actual por lo que no se menciona en los siguientes apartados.
Recomendaciones para el diagnóstico de la epidermólisis bullosa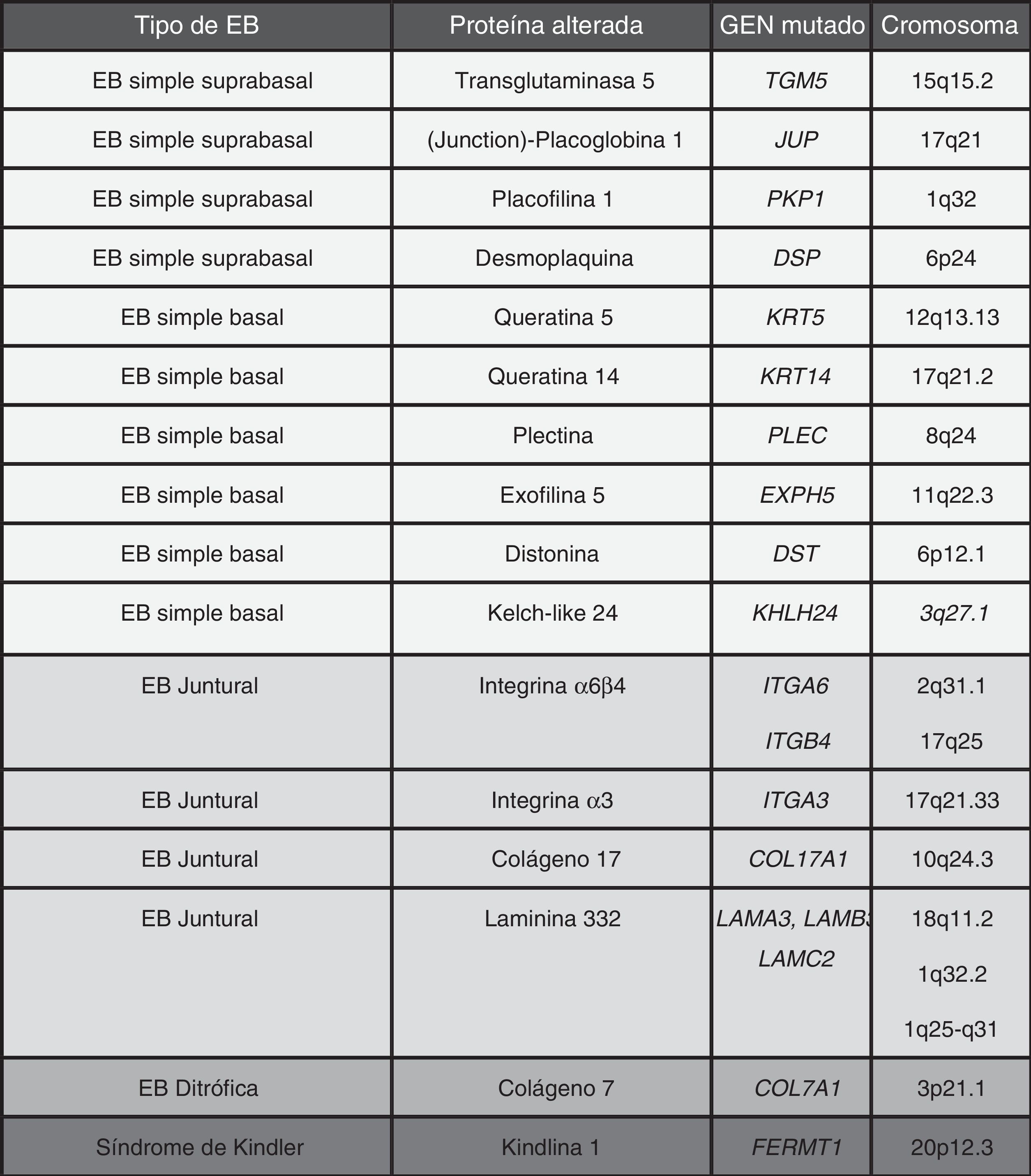
Dado el gran número de proteínas involucradas en esta enfermedad, el diagnóstico de la EB es complejo y su clasificación ha estado sujeta, durante los últimos 20 años, a revisiones internacionales consensuadas periódicas, la última en 20132. La última clasificación de la EB se basa en un sistema de capas (onion skinning) que tiene en cuenta, de modo secuencial, el nivel de escisión de la piel, las características fenotípicas, el modo de herencia, los hallazgos en el mapeo antigénico, el gen implicado y la mutación causal de la enfermedad2. Siguiendo estos criterios se han descrito hasta 30 subtipos de EB.
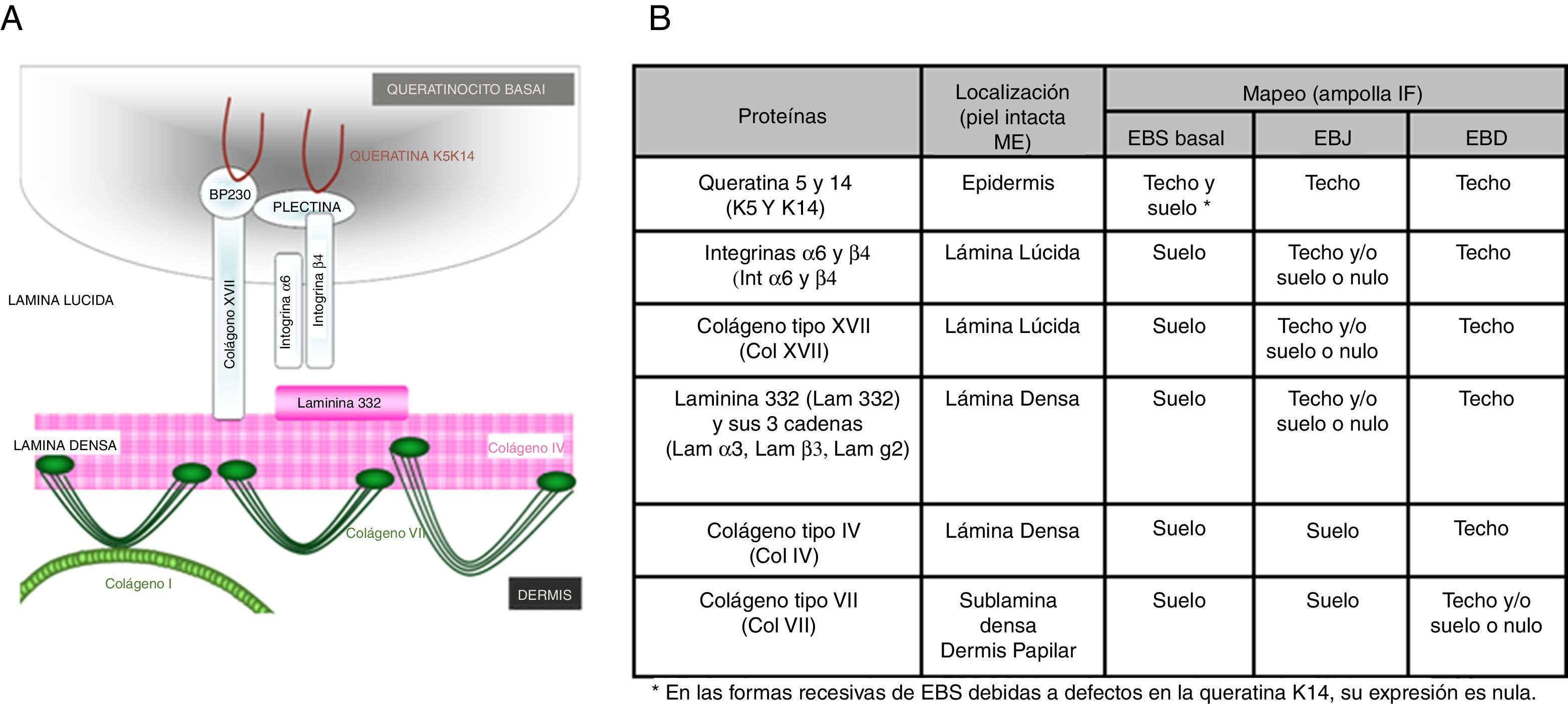
Frente a la mencionada complejidad, el diagnóstico de la EB implica, además de la obtención de una muestra de sangre para extraer el ADN genómico, la toma de una biopsia cutánea que incluya una zona de despegamiento entre la dermis y la epidermis que se induce por fricción en el momento de la recolección. Esta biopsia se emplea para realizar un mapeo antigénico que permita establecer el plano de rotura y que oriente el diagnóstico hacia uno de los 4principales tipos de EB y, por tanto, que acote el gen candidato. Para el mapeo antigénico se emplea habitualmente una batería de anticuerpos dirigidos contra el colágeno IV (Col iv) y distintos antígenos presentes en las proteínas de la membrana basal implicadas en las formas más comunes de EB, cuya expresión se analiza por inmunofluorescencia. En muchas formas recesivas de EB, las mutaciones de pérdida de función generalmente conducen a una reducción o ausencia del inmunomarcaje para una proteína en particular, lo que hace de este modo posible la identificación del mejor gen candidato. Sin embargo, en general, en las formas dominantes (y en algunas recesivas), los resultados del mapeo antigénico pueden no mostrar diferencias claras en el nivel de expresión de las distintas proteínas analizadas. En estos casos es fundamental que la muestra contenga una ampolla reciente para poder determinar el plano de fractura. Cabe resaltar que la microscopia electrónica de trasmisión empleada inicialmente en el diagnóstico ha sido casi reemplazada por el mapeo antigénico.
Finalmente, la identificación de la mutación —en el gen que codifica la proteína cuya alteración da lugar a la enfermedad— permite la realización de un diagnóstico definitivo, ayuda —dentro de ciertos límites— a realizar un pronóstico de la evolución de la enfermedad y es clave para la inclusión del paciente en ensayos clínicos. Existen protocolos estándar que incluyen reacción en cadena de la polimerasa y posterior secuenciación Sanger. Para analizar los 18 genes implicados en los distintos subtipos de EB se requieren más de 400 pares de cebadores, de ahí la conveniencia de acotar los genes candidatos mediante el mapeo antigénico de las biopsias de piel. La búsqueda de la mutación es muy laboriosa, ya que se han descrito más de 1.000 mutaciones y, en muchos casos, se siguen encontrando mutaciones nuevas asociadas únicamente a una familia. Debido a limitaciones de la técnica de secuenciación directa, las mutaciones patogénicas no son identificadas en alrededor del 15% de los casos. La incorporación de la tecnología de secuenciación masiva, next generation sequencing (NGS), permite en la actualidad mejorar la sensibilidad en el diagnóstico de EB16.
En los muchos casos en los que la mutación identificada resulta ser «privada» de cada familia (no ha sido encontrada previamente en otro paciente), es necesario asegurarse de que no se trata de una variante polimórfica no patogénica. Para ello, debe investigarse su presencia y frecuencia en la población general del mismo origen. Además, es conveniente constatar el efecto patogénico de la mutación a nivel del ARN mensajero o de la proteína. Para dichos estudios se emplea una segunda pequeña biopsia de piel de la que se aislarán las células de la dermis y la epidermis del paciente, que se recomienda recoger en el mismo acto quirúrgico que la empleada para el inmunomapeo. Los datos recabados serán de interés para establecer la estrategia de estudio en el laboratorio, con el fin último de proporcionar al paciente y sus familiares un adecuado consejo o asesoramiento genético para cada situación en particular. Una vez que se ha informado al paciente y a sus familiares en el contexto de esta consulta es cuando realmente finaliza el diagnóstico.
En la tabla 2 se recogen las recomendaciones sobre los pasos para el diagnóstico de la EB.
Pasos recomendados frente a la sospecha de EB
| I. Diagnóstico clínico |
| II. Diagnóstico molecular |
| Inmunomapeo antigénico: selección del gen candidato |
| Diagnóstico genético: detección de mutaciones por PCR y secuenciación |
| III. Asesoramiento o consejo genético |
| Podrán, además, derivarse las siguientes acciones de intervención médica: |
| IV. Diagnóstico prenatal (invasivo y no invasivo) |
| V. Diagnóstico preimplantacional |
El diagnóstico de la EB se inicia con la realización de una historia clínica completa del paciente que recoge las manifestaciones cutáneas y extracutáneas de la enfermedad, el momento de aparición de los síntomas, e incluye antecedentes familiares de interés. El árbol genealógico debe reflejar, entre otras cosas, la edad de los padres, la posible consanguinidad y el origen geográfico de la familia.
En el neonato, el diagnóstico clínico del subtipo de EB es complicado, ya que muchos de los síntomas iniciales de la enfermedad son comunes a los 4tipos principales, lo que hace prácticamente imposible discernir entre ellos. Es, además, fundamental realizar el diagnóstico diferencial entre las formas hereditarias de la EB y las enfermedades ampollosas autoinmunes17,18. Estas últimas se deben a lesiones causadas por la acción inflamatoria de autoanticuerpos que reaccionan contra determinadas moléculas de la unión dermoepidérmica y no a mutaciones en el gen que codifica para dichas proteínas, como ocurre en las formas hereditarias. La existencia de antecedentes familiares aporta, sin duda, información muy valiosa.
En los pacientes adultos, en los que la sintomatología de la enfermedad está completamente establecida, las manifestaciones pueden llegar a ser muy específicas y permiten orientar el diagnóstico hacia alguno de los subtipos. En la última clasificación2 se estratifican por niveles, cada rasgo clínico en cada subtipo de EB, su intensidad, momento de aparición, etc. Esta información es muy útil para diferenciar entre algunos subtipos con características comunes2. No obstante, en las formas leves de EB los subtipos pueden seguir siendo, en muchos casos, clínicamente indistinguibles en el adulto.
Presentamos a continuación un resumen de las principales características de cada tipo y subtipo de EB, algunas imágenes clínicas orientativas y la tabla 3, donde consultar las proteínas afectas en cada subtipo.
Clasificación clínica de la EB
| Tipo | Subtipo | Subtipo clínico | Proteína |
|---|---|---|---|
| EBS | |||
| Suprabasal | |||
| Síndrome de descamación de la piel acral (APSS) | Trasglutaminasa 5 | ||
| Superficial (EBSS) | Desconocida | ||
| Acantolítica (EBS-acanth) | Desmoplaquina, placoglobina | ||
| Síndromes de fragilidad cutánea: | |||
| - EBS-desmoplaquina: síndrome de fragilidad cutánea-pelo lanoso | Desmoplaquina | ||
| - EBS-placoglobina | Placoglobina | ||
| - EBS-placofilina: síndrome de fragilidad cutánea-displasia ectodérmica | Placofilina1 | ||
| Basal | |||
| Localizada (EBS-loc) | Keratina 5 (K5), keratina 14 (K14) | ||
| Grave generalizada (EBS-gen sev) | Keratina 5 (K5), keratina 14 (K14) | ||
| Generalizada intermedia (EBS-gen intermed) | Keratina 5 (K5), Keratina 14 (K14) | ||
| Con pigmentación moteada (EBS-MP) | Keratina 5 (K5) | ||
| Circinada migratoria (EBS-migr) | Keratina 5 (K5) | ||
| Autosómica recesiva K14 (EBS-AR K14) | Keratina 14 (K14) | ||
| Con distrofia muscular (EBS-MD) | Plectina | ||
| Con atresia pilórica (EBS-PA) | Plectina, Integrina a6b4 | ||
| Ogna (EBS-Og) | Plectina | ||
| Autosómica recesiva-deficiencia de BP230 (EBS-AR BP230) | Antigeno 1 pénfigo bulloso (BP230) | ||
| Autosómico recesivo-deficiencia de exofilina 5 (EBS-AR exofilina 5) | Exofilina 5 | ||
| EBJ | |||
| Generalizada | |||
| Grave generalizada (EBJ-sev gen) | Laminina 332 | ||
| Generalizada intermedia (EBJ-gen intermed) | Laminina 332, colágeno XVII | ||
| Con atresia pilórica (EBJ-PA) | Integrina α6β4 | ||
| Localizada | |||
| Localizada (EBJ-loc) | Laminina 332, colágeno XVII, integrina b4 | ||
| Inversa (EBJ-inv) | Laminina 332 | ||
| Síndrome de LOC | Laminina 332, cadena α3 | ||
| EBD | |||
| Dominante | |||
| Generalizada (EBDD-gen) | Colágeno VII | ||
| Localizada (EBDD-loc) | Colágeno VII | ||
| Acral (EBDD-ac) | Colágeno VII | ||
| Pretibial (EBDD-pt) | Colágeno VII | ||
| Pruriginosa (EBDD-pr) | Colágeno VII | ||
| Solo uñas (EBDD-na) | Colágeno VII | ||
| Del recién nacido (EBDD-BDN) | Colágeno VII | ||
| Recesiva | |||
| Grave generalizada (EBDR-gen sev) | Colágeno VII | ||
| Generalizada intermedia (EBDR-gen intermed.) | Colágeno VII | ||
| Inversa (EBDR-inv) | Colágeno VII | ||
| Localizada (EBDR-loc) | Colágeno VII | ||
| Pretibial (EBDR-pt) | Colágeno VII | ||
| Pruriginosa (EBDR-pr) | Colágeno VII | ||
| Centrípeta (EBDR-ce) | Colágeno VII | ||
| Del recién nacido (EBDR-BDN) | Colágeno VII | ||
| SK | |||
| Kindlina 1 |
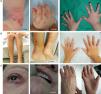
El término EBS incluye a todas las formas de EB en las que la fragilidad cutánea está confinada a la epidermis. En la gran mayoría de las EBS la fractura se produce a nivel histológico en el estrato basal y la enfermedad se transmite de forma autosómica dominante. La herencia autosómica recesiva en EBS representa solo el 5% de los casos. Aunque las manifestaciones clínicas (fig. 2) comienzan, en general, al nacimiento o en el periodo neonatal, en algunos casos pueden aparecer en la edad adulta. Como norma general, las ampollas en la EBS se inducen por traumatismo y, raramente, de forma espontánea. Las cicatrices, los quistes de milia y la distrofia ungular suelen ser menos frecuentes en la EBS que en la EBJ y la EBD.
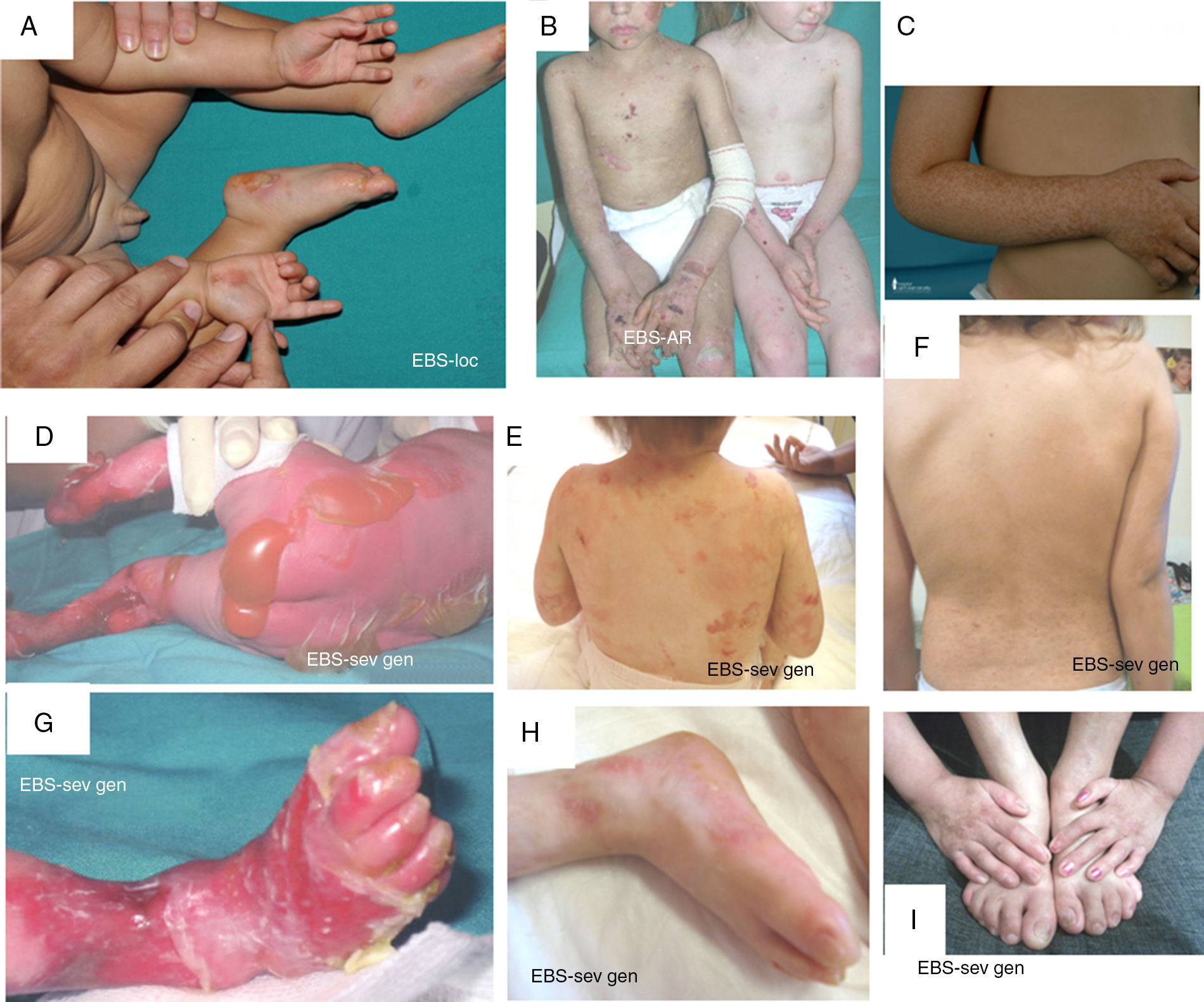
Manifestaciones clínicas de la EBS basales. Los genes de las queratinas (KRT5, KRT14) son los causales de la enfermedad en todos los pacientes19,20. Fotografías obtenidas con consentimiento informado expreso. (Fuente: u714-CIBERER-CIEMAT-UC3M-IISFJD).
A) Afectación palmoplantar de un paciente con EBS-localizada durante la infancia. B) Afectación generalizada de 2hermanos con EBS, de padres consanguíneos, con un patrón de herencia autosómica recesiva19 (arriba). C) Pigmentación moteada en un paciente con EBS-PM21. D-G) Amplia afectación de un neonato con EBS sev gen. E-F-H-I) Progresión durante la infancia. E) Se pueden observar las lesiones herpetiformes ampliamente distribuidas en la espalda.
La EBS localizada es la forma más común y se caracteriza por ampollas confinadas a las palmas de las manos y las plantas de los pies y, en algunos de los casos, por erosiones o ampollas orales durante la infancia. También pueden presentarse lesiones en cualquier otra parte del cuerpo como consecuencia de traumatismos cutáneos. La EBS grave generalizada, un subtipo menos frecuente, suele estar asociada a una marcada morbimortalidad en el período neonatal o infancia temprana. Su rasgo más distintivo es la formación de vesículas intactas agrupadas con una distribución en forma de arco (herpetiforme). Estos pacientes suelen desarrollar un engrosamiento de la piel en palmas y plantas que da lugar a una queratosis que puede resolverse en la edad adulta. Algunos pacientes presentan distrofia ungular, cicatrices atróficas, quistes de milia, afectación de las mucosas (estenosis laríngea), anemia y retraso en el crecimiento. A diferencia de la forma grave, la EBS generalizada intermedia presenta ampollas no herpetiformes y en ella la anemia y el retraso en el crecimiento son infrecuentes2.
Otros subtipos de EBS basales menos frecuentes son la de pigmentación moteada, circinada migratoria, autosómica recesiva (EBS-AR), con distrofia muscular, con atresia pilórica, ogma, déficit de BP230 y exofilina 52. Recientemente, se ha identificado una nueva proteína (kelch-like 24), asociada a un subtipo de EBS basal con características clínicas distintivas13 con respecto a las formas clásicas de EBS (alopecia, atrofia folicular y cutánea).
Actualmente, el término EBS engloba también a aquellas formas en las que la fragilidad mecánica ocurre, a diferencia de las descritas anteriormente, en los estratos suprabasales. Este subtipo de EBS suprabasal incluye a la forma acral, superficial, acantolítica y síndromes de fragilidad cutánea debidos a deficiencias de desmoplaquina, placoglobina y placofilina4 (ver tablas 1 y 3).
Epidermólisis bullosa junturalLa característica clínica más señalada en todos los subtipos de EBJ es la hipoplasia del esmalte, rasgo distintivo útil para el diagnóstico de este tipo de EB (fig. 3F). La EBJ también se subdivide en varios subtipos que se transmiten, todos ellos, de forma autosómica recesiva.
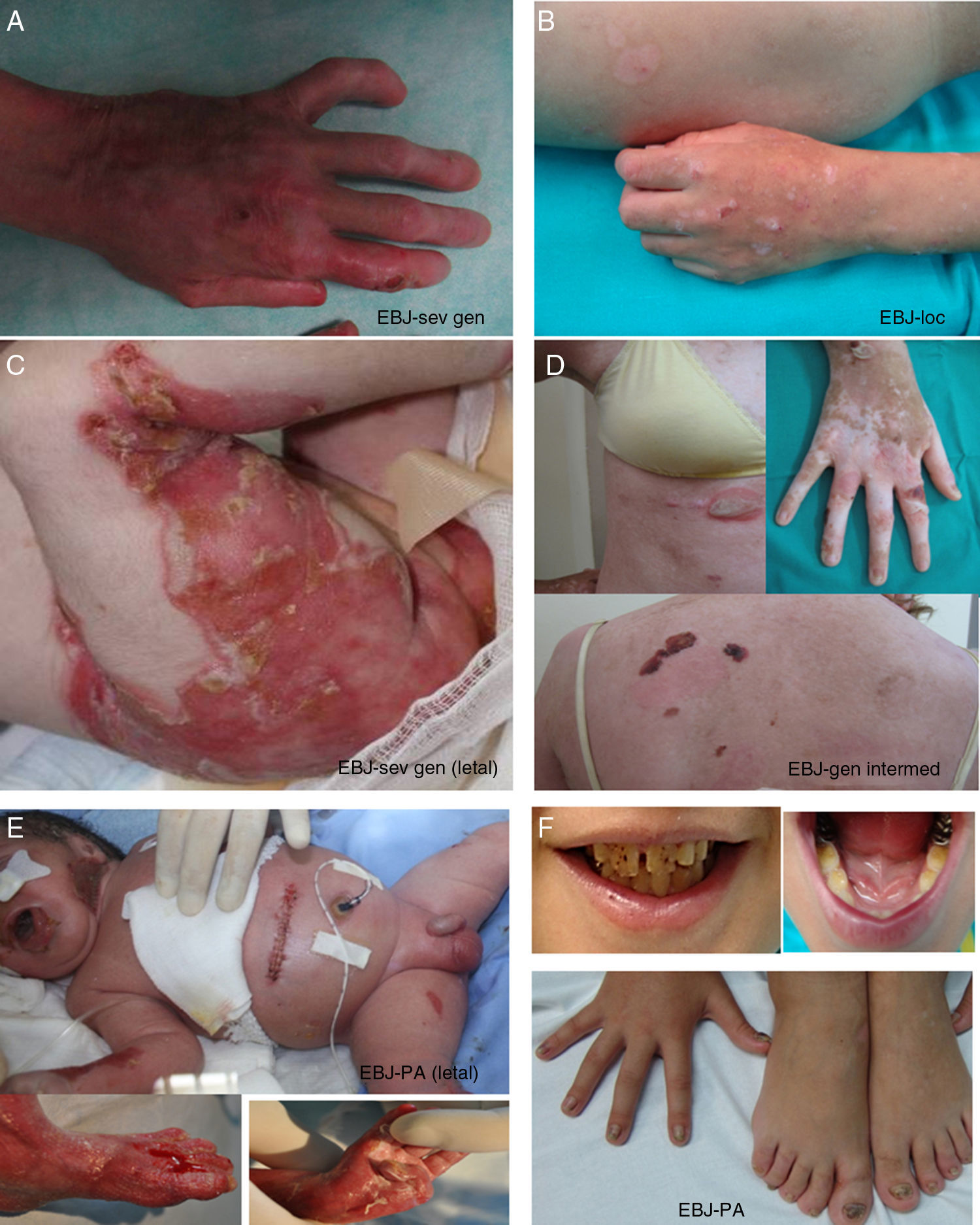
Manifestaciones clínicas de la EBJ. Fotografías obtenidas con consentimiento informado expreso (Fuente: U714-CIBERER-CIEMAT-UC3M-IISFJD). A-B) EBJ grave generalizada causada por mutaciones en el gen LAMC2 con ausencia de Lam 332, concretamente la cadena γ2, que causó la muerte en el periodo neonatal a uno de los pacientes. C) EBJ-generalizada intermedia. D) EBJ-localicada. En ambos pacientes se encontraron mutaciones en el gen COL17A1, que resultaron en una expresión reducida y en la ausencia completa de Col17, respectivamente. Nótese la hipopigmentación postinflamatoria. E-F) EBJ con atresia de píloro causada por mutaciones en el gen ITGB4; la expresión de la proteína fue comparable a la de un control en el paciente F y nula en la forma letal22. Ambos pacientes fueron tratados mediante cirugía correctora de la atresia pilórica.
El subtipo más grave es la EBJ grave generalizada, que aparece desde el nacimiento y puede afectar a toda la extensión de la piel (fig. 3 A-B). Un hallazgo muy significativo de este subtipo es la aparición de tejido de granulación exuberante de distribución simétrica, en la boca, la región central de la cara y nariz, la región superior de la espalda, axilas y los pliegues inguinales. El cuadro clínico incluye ampollas intraorales, microstomía y anquiloglosia, estas últimas no tan marcadas como en las formas recesivas de EBD. Las manifestaciones extracutáneas pueden, además, afectar a otros órganos como el esófago, los ojos, la vía aérea superior y los genitales. Los pacientes presentan también retraso en el crecimiento y anemia multifactorial. Existe un alto riesgo de mortalidad infantil (hasta los 2años de vida) como resultado del retraso del crecimiento, infecciones, neumonía y obstrucción de las vías aéreas.
La EBJ generalizada intermedia, evidente desde el nacimiento, es la más común de las junturales, con un trastorno generalizado que cursa con ampollas, cicatrices atróficas y uñas distróficas o ausentes (fig. 3D). Los pacientes suelen presentar, además, hipopigmentación postinflamatoria o despigmentación y, en algunos de los casos, alopecia cicatricial del cuero cabelludo. El riesgo de muerte por oclusión de la vía aérea o por otras complicaciones es mucho menor que en la grave generalizada, al igual que la anemia y el retraso en el crecimiento. No obstante, algunos de los casos pueden desarrollar carcinomas epidermoides.
Otro subtipo clínicamente importante es la EBJ inversa, que se presenta con ampollas graves y confinadas a lugares de la piel intertriginosos, esófago y vagina. El síndrome de LOC se caracteriza por la formación de ampollas y cicatrices localizadas en cara y cuello con tejido de granulación, alteraciones en las vías respiratorias superiores, anormalidades en las uñas e hipoplasia del esmalte dental2. La EBJ con atresia pilórica cursa con ampollas generalizadas desde el nacimiento y atresia congénita del píloro y ocasiona anomalías congénitas del tracto genitourinario con un pronóstico muy variable: puede causar muerte infantil neonatal o dar lugar a un fenotipo más leve (fig. 3 E-F). Cabe mencionar que existe una forma de EB asociada a atresia de píloro en la que el plano de fractura cutánea ocurre en el estrato epidérmico basal y no en la unión dermoepidérmica y que se considera, por tanto, un subtipo de EBS (EBS con atresia pilórica; tablas 1 y 3).
Epidermólisis bullosa distróficaLa EBD se subdivide en 2grandes grupos, dominante y recesiva, en función del tipo de herencia (tabla 3, fig. 4)1,2.
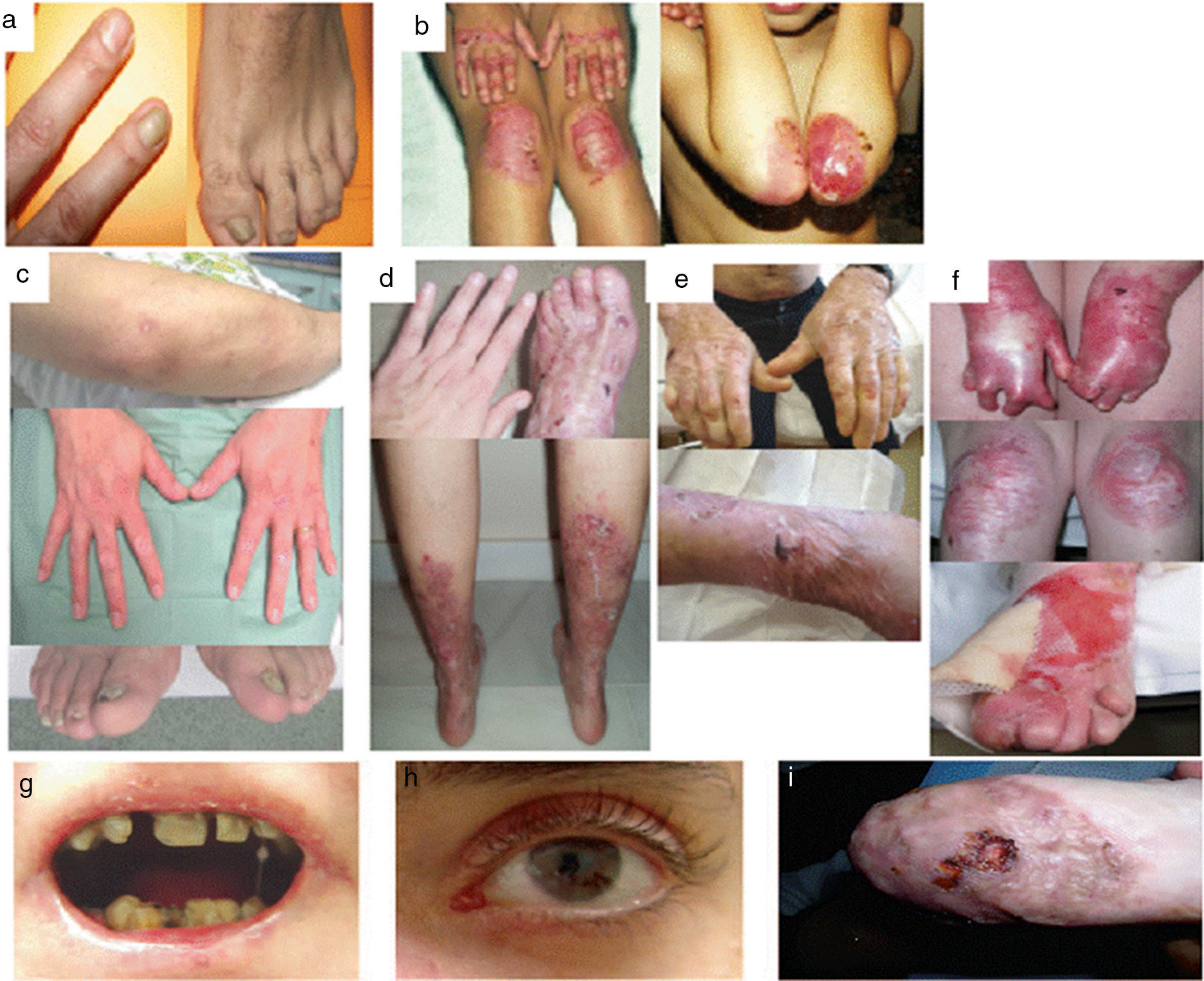
Manifestaciones clínicas de la EBD. Fotografías clínicas de pacientes españoles obtenidas con consentimiento informado expreso (Fuente: U714-CIBERER-CIEMAT-UC3M-IISFJD). El gen que codifica para el colágeno de tipo VII (COL7A1) es el causante de la enfermedad en todos los pacientes23,24). a) EBD dominante, solo uñas. b) EBD dominante, generalizada. c) EBD recesiva, localizada. d) EBD recesiva, pretibial. e) EBD recesiva, generalizada (no Hallopeau-Siemens). f) EBD recesiva, grave generalizada (Hallopeau-Siemens). g-i) Otras complicaciones en pacientes con EBDR-sev gen. g) Microstomía con alteraciones dentales y caries. h) Cicatrices y ampollas oculares (párpado inferior). i) Carcionama escamoso celular en el muñón de la mano izquierda.
El subtipo más grave de la EBD dominante (EBDD) es el generalizado, que se caracteriza por la presencia de ampollas distribuidas por todo el cuerpo desde el nacimiento que, con el tiempo, se asocian con quistes de milia, atrofia, cicatrices y uñas distróficas (fig. 4B). Es frecuente la aparición de ampollas y erosiones recurrentes en el esófago. Otros subtipos menos graves de EBDD son el de solo uñas (fig. 4A), el acral, la forma de EBDD localizada con ampollas confinadas en manos y pies (fig. 4C), la pretibial, que además cursa con lesiones menores en forma de placa, a menudo violáceas (fig. 4D), la forma pruriginosa, que da lugar a un prurito intratable, la del recién nacido, con ampollas generalizadas que van acompañadas de cicatrización focal y que suele desaparecer entre los 6 y los 24 meses de vida.
La EBD recesiva (EBDR), que a su vez se subdivide en varios grupos, puede exhibir un amplio espectro de gravedad, que va desde una simple distrofia ungular hasta la presencia generalizada de ampollas y deformidades en manos y pies (pseudosindactilia). La enfermedad puede afectar a las mucosas y, en las formas más graves, se asocia a un alto riesgo de desarrollar carcinomas epidermoides. El subtipo más frecuente es el grave generalizado, que es evidente en el nacimiento, con ampollas que van extendiéndose por todo el cuerpo de forma progresiva y resultan en cicatrización atrófica. Este subtipo cursa con tejido de granulación, sobre todo en las heridas crónicas, así como contractura articular y desarrollo de pseudosindactilia (fig. 4F). Los pacientes suelen desarrollar, además, caries en exceso, anquiloglosia grave y microstomía (fig. 4G) asociadas a un déficit en la ingesta de alimentos. Otras manifestaciones extracutáneas son las ampollas corneales, estenosis esofágica, insuficiencia renal crónica, anemia multifactorial y retraso en el crecimiento. Este subtipo, que es el más grave de EBDR, se complica habitualmente con el desarrollo de carcinomas epidermoides metastásicos (fig. 4I) y muerte prematura del paciente. El subtipo generalizado intermedio es menos grave (fig. 4E), no cursa, en general, con retraso en el crecimiento ni anemia, la frecuencia de aparición de estenosis esofágica es menor y la afectación cutánea, también. Sin embargo, sigue teniendo un riesgo significativo de desarrollar carcinomas de células escamosas. Subtipos menos comunes son el centrípeto, que empieza en la zona acral y se va extendiendo por el tronco, y el inverso (EBDR-inv), en el que la aparición de ampollas está restringida a las zonas intertriginosas, la base del cuello, la parte alta de la espalda y la zona lumbosacra. Los pacientes con EBDR-inv son propensos a desarrollar ampollas en la cavidad oral, esófago y tracto genitourinario. Pueden llegar a desarrollar estenosis debilitante en esófago y vagina.
Síndrome de KindlerEl SK es una genodermatosis con un patrón de herencia autosómico recesivo que está incluido en la clasificación de EB con base en criterios clínicos y biológicos, lo que permite que estos pacientes (menos de 200 individuos en el mundo) sean atendidos en los centros de referencia de EB1,2. El SK aparece al nacimiento con la presencia de ampollas que pueden distribuirse de forma generalizada y, frecuentemente, desaparecen con la edad (fig. 5). El curso de esta enfermedad se caracteriza por el desarrollo progresivo de poiquiloderma, fotosensibilidad, envejecimiento cutáneo prematuro y queratoderma25. Los pacientes con SK pueden presentar cicatrización atrófica, distrofia de uñas, contractura y sinequia interdigital. Complicaciones como colitis grave, esofagitis, estenosis uretral, hiperplasia gingival y carcinomas escamosos pueden verse asociadas al SK1,2. El estado prooxidativo, recientemente descrito en las células epiteliales de pacientes con SK, podría justificar las características patognomónicas no asociadas a fragilidad cutánea26,27.
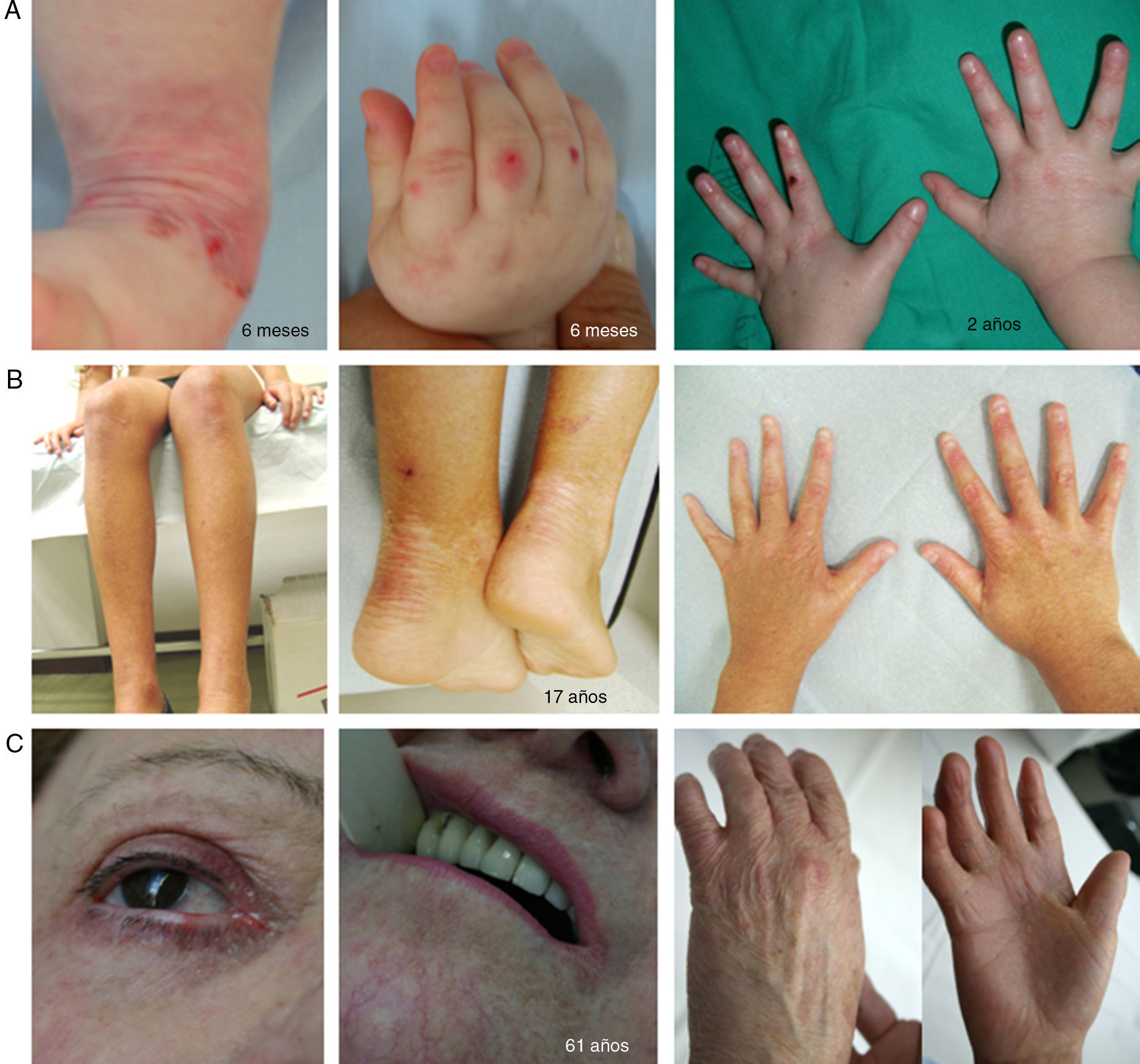
Manifestaciones clínicas del SK. Fotografías clínicas de pacientes obtenidas con consentimiento informado expreso (Fuente: U714-CIBERER-CIEMAT-UC3M-IISFJD). El gen que codifica para la kindlina (KIND1) es el causante de la enfermedad en todos los pacientes26-28. A) Erosiones y envejecimiento prematuro de la piel de los dorsos de las manos incipiente en la infancia. B) Envejecimiento cutáneo y poiquiloderma evidentes junto con distrofia ungueal en una paciente joven. C) Afectación ocular, poiquiloderma, sinequia interdigital y contractura en las manos de una paciente sexagenaria.
El primer paso recomendado para el diagnóstico de la EB es la realización de una clasificación clínica de los pacientes con un cuadro de fragilidad cutánea que nos hace sospechar una posible alteración en las proteínas de anclaje dermoepidérmico14,15. El diagnóstico continúa con la clasificación inicial dentro de los 4tipos principales de EB mediante la identificación del nivel de rotura de la piel por inmunofluorescencia (tabla 4).
Plano de fractura en cada uno de los 4principales tipos de EB
| Principales tipos de EB | Nivel de formación de las ampollas |
|---|---|
| EB simple | Intraepidérmica |
| EB juntural | Intralámina lúcida (membrana basal) |
| EB distrófica | Sublámina densa (dérmica) |
| Síndrome de Kindler | Múltiples niveles (intraepidérmica, intralámina lúcida y sublámina densa) |
Para el inmunomapeo antigénico, se emplean cortes histológicos obtenidos a partir de biopsias de piel criopreservadas que contienen una ampolla inducida por fricción en el momento de la toma de la muestra. Estos cortes se incuban con una batería de anticuerpos dirigidos contra el Col IV y distintos antígenos de proteínas implicadas en EB. El anticuerpo contra Col IV, proteína presente en la lámina densa de la unión dermoepidérmica, nos permite identificar de forma inequívoca la EBD (localización del marcaje en el techo de la ampolla) y el SK (reduplicación de la membrana basal). El uso de este anticuerpo no distingue entre la EBS y la EBJ, ya que en ambos casos el marcaje se localiza en el suelo de la ampolla (fig. 6).
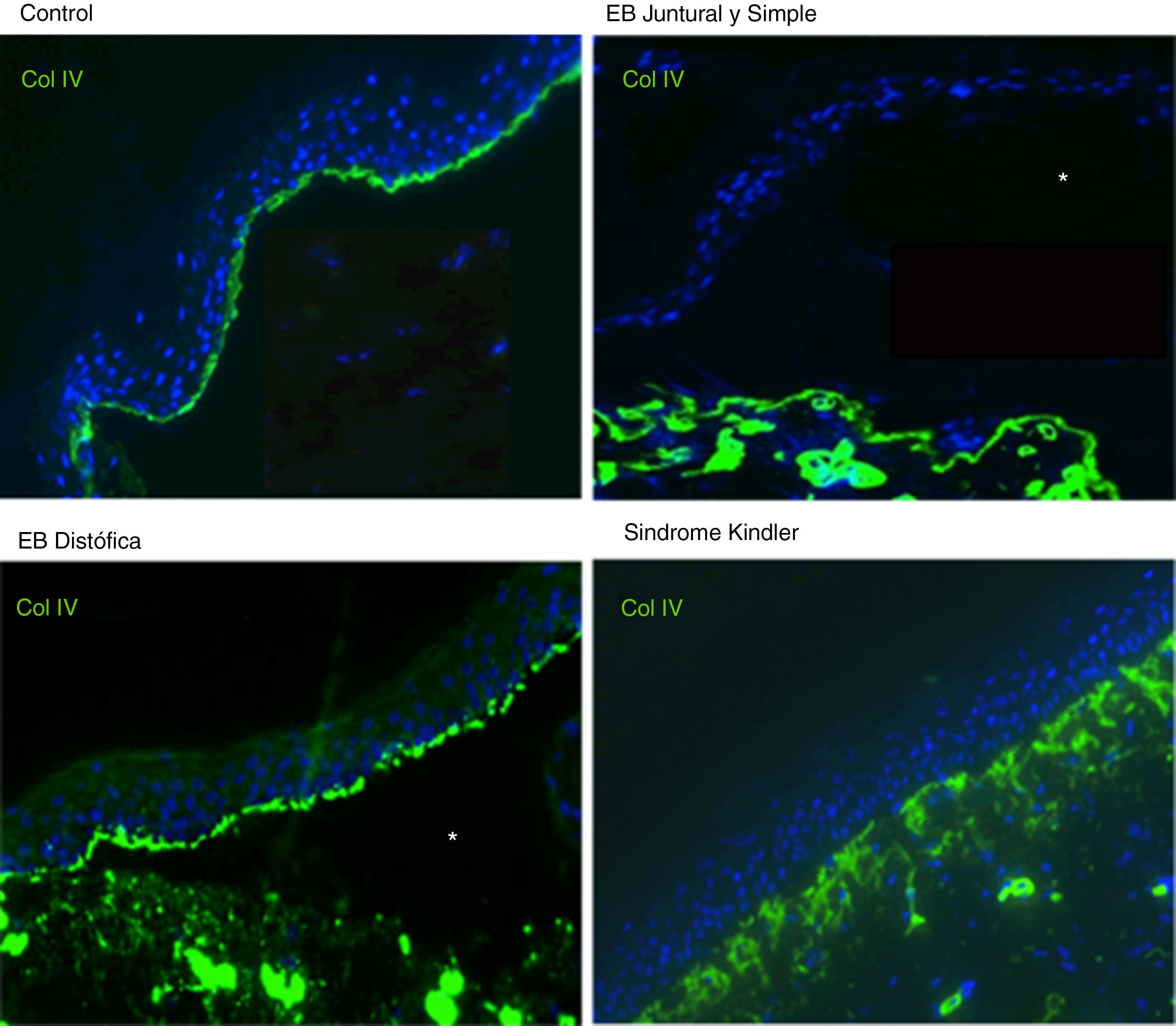
Inmunofluorescencia Col IV. Tanto en la EBJ como en la EBS puede observarse el Col IV en el suelo de la ampolla. En la EB distrófica se observa el Col IV en el techo de la ampolla, mientras que en el SK se observa una reduplicación de la membrana basal distintiva de esta enfermedad. Fuente: Dra. Marta García (U714-CIBERER-CIEMAT-UC3M-IISFJD).
El uso de anticuerpos específicos para las proteínas implicadas en EB y su patrón de expresión en el techo y el suelo de la ampolla nos permite, además, diferenciar entre EBS, EBJ y EBD (figs. 7 y 8) e ir acotando el gen candidato.
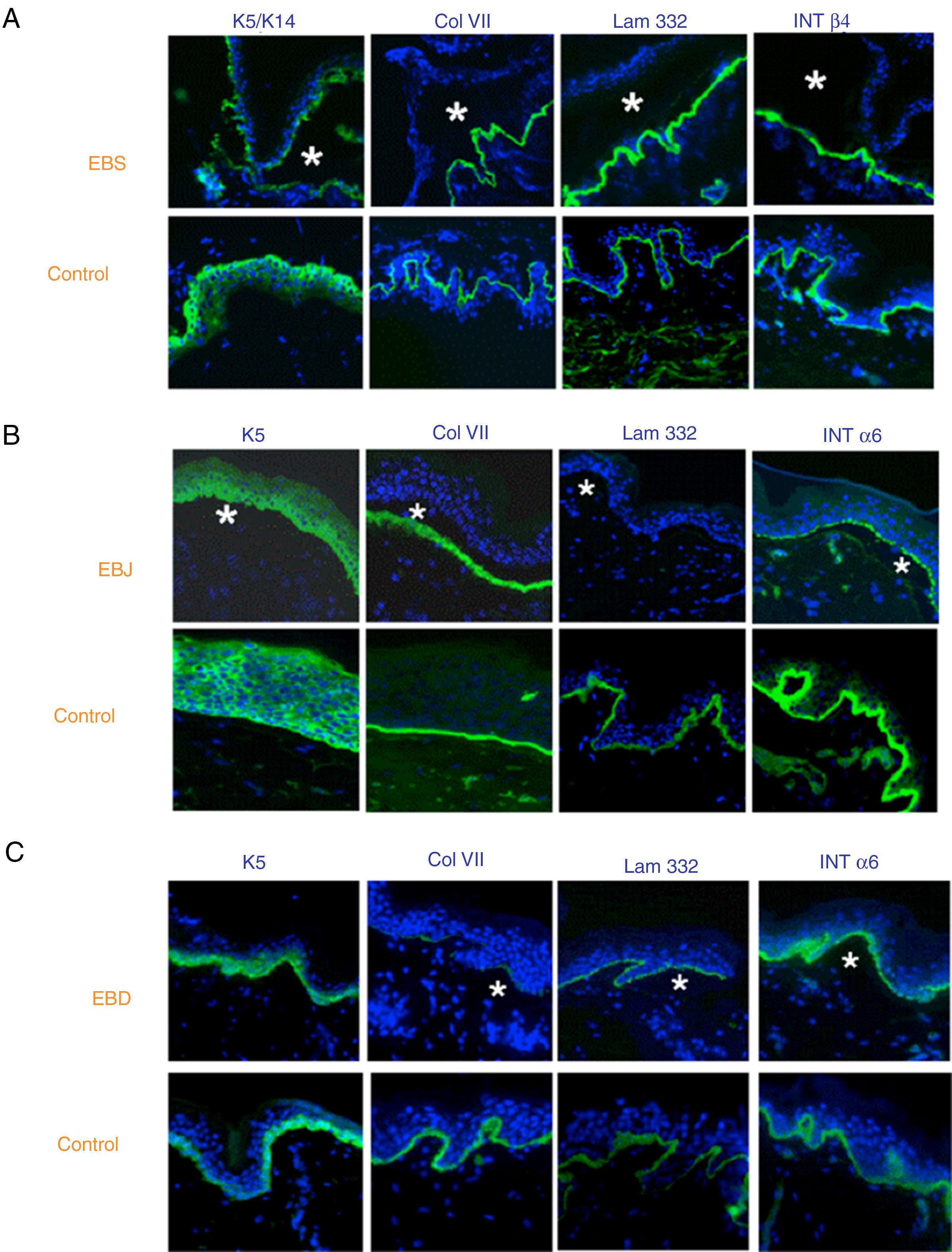
Clasificación de la EB y selección del gen candidato mediante mapeo antigénico (primer cribado) en cortes histológicos de biopsias de piel criopreservadas. Microfotografías: Dra. Marta García (U714-CIBERER-CIEMAT-UC3M-IISFJD). A) Inmunofenotipo de la EBS basal. Las queratinas K5 y K14 marcan el suelo y el techo de la ampolla como resultado de una fractura intraepidérmica. El marcaje para el resto de las proteínas las localiza en el suelo de la ampolla. Este resultado nos indica que los genes candidatos en el primer cribado son KRT5 y KRT14. B) Inmunofenotipo de la EBJ. Como resultado de una fractura intralámina lúcida el marcaje de las queratinas queda en el techo de la ampolla, el de colágeno VII queda en el suelo de la ampolla. En este caso, la ausencia de marcaje de laminina 332 nos indica que los genes candidatos son LAMA3, LAMB3 o LAMC3 y la subclasificación molecular de EBJ (ver tabla 5). Se observa, además, que la ausencia en Lam332 puede afectar a la expresión de las integrinas. C) Inmunofenotipo de la EBD. Como resultado de una fractura a nivel de la dermis papilar, todos los marcadores mapean en el techo de la ampolla. Este resultado nos indica que el gen candidato es el COL7A1. La práctica ausencia de Col VII es consistente con la EBD grave generalizada (ver tabla 5).
En algunos casos, los resultados de la inmunofluorescencia permiten afinar aún más en la subclasificación, ya que la disminución de la intensidad de la tinción (tabla 5) o la ausencia total de marcaje de una proteína en particular señalan de forma consistente al gen que la codifica como candidato. La ausencia completa de marcaje se corresponde generalmente con mutaciones nulas. En otros casos se producen proteínas completas con la secuencia de aminoácidos alterada y su expresión puede llegar a ser comparable a la de un individuo sano. El proceso de selección del gen candidato se especifica en la figura 7.
Clasificación molecular de la EB
| Tipo | Subtipo | Proteína | Expresión prot. | Gen | |
|---|---|---|---|---|---|
| EBS | |||||
| Suprabasal | Trasglutaminasa 5 | Normal, reducida o ausente | TGM5 | ||
| Plakofilina 1 | Reducida o ausente | PKP1 | |||
| Desmoplakina | Reducida o ausente | DSP | |||
| Placoglobina | Reducida o ausente | JUP | |||
| Basal | Keratina 5 | Usualmente normal | KRT5 | ||
| Keratina 14 | Usualmente normal | KRT14 | |||
| Plectina | Reducida o ausente | PLEC | |||
| Exofilina 5 (slac2-b) | Ausente | EXPH5 | |||
| Antígeno penfigoide bulloso1 (BPAG1; BP230) | Ausente | DST | |||
| EBJ | |||||
| Generalizada | |||||
| Grave generalizada (gen sev) | Laminina 332 | Muy reducida o ausente | LAMA3, LAMB3, LAMC2 | ||
| Generalizada intermedia (gen intermed) | Laminina 332 | Reducida | LAMA3, LAMB3, LAMC2 | ||
| Colágeno XVII | Reducida o ausente | COL17A1 | |||
| Con atresia pilórica (PA) | Integrina α6β4 | Muy reducida o ausente | ITGB4, ITGA6 | ||
| Con inicio tardío (LO) | Colágeno XVII | Reducida o patrón anormal | COL17A1 | ||
| Con afectación respiratoria y renal (RR) | Subunidad integrina a3 | Normal o ausente | ITGA3 | ||
| Localizada | |||||
| Localizada (Loc) | Colágeno XVII | Reducida o ausente | COL17A1 | ||
| Laminina 332 | Reducida | LAMA3, LAMB3, LAMC2 | |||
| Integrina a6b4 | Reducida | ITGB4 | |||
| Inversa (Inv-I) | Laminina 332 | Reducida | LAMA3, LAMB3, LAMC2 | ||
| Síndrome de LOC | Laminina 332, cadena isoforma a3 | Normal | LAM3A | ||
| EBD | |||||
| Dominante (EBDD) (todos los tipos excepto el del recién nacido) | Colágeno VII | Normal o reducido | COL7A1 | ||
| Dominante y Recesivo (del recién nacido) | Colágeno VII | Tinción granular dentro de queratinocitos basales y reducido o ausente en la unión dermoepidérmica cuando la enfermedad está activa; tinción normal en la unión dermoepidérmica en adultos y se vuelve asintomática | COL7A1 | ||
| Recesiva (EBDR) | |||||
| Grave generalizada | Colágeno VII | Ausente o muy reducida | COL7A1 | ||
| Intermedia generalizada | Colágeno VII | Reducida | COL7A1 | ||
| Localizada | Colágeno VII | Reducida o normal | COL7A1 | ||
| Otras | Colágeno VII | Reducida | COL7A1 | ||
| Kindler | |||||
| Kindlina 1 | FERMT1-KIND1 |
Este primer cribado diagnóstico mediante inmunofenotipado, gracias a la secuenciación masiva, no es hoy por hoy imprescindible, pero sí es recomendado como paso previo al análisis genético. El inmunofenotipado ayuda en la identificación de la proteína implicada y, por tanto, del gen candidato23,29,30. En la tabla 5 se muestran los diferentes subtipos de EB con base en criterios moleculares: proteínas afectadas, nivel de expresión en piel y los genes que las codifican.
Diagnóstico genéticoTras el conocimiento de la proteína afectada en el paciente, podemos comenzar el análisis genético mediante la secuenciación del gen candidato. Se inicia con la búsqueda de las mutaciones más comunes y finaliza con la secuenciación completa del gen, si fuese necesario. Actualmente, la búsqueda de la mutación se realiza por secuenciación Sanger, aunque ya se están incorporando técnicas nuevas, como la NGS, que pueden incluir la búsqueda de mutaciones en diferentes genes al mismo tiempo16. Este avance es muy útil, ya que permite realizar el estudio molecular directo independizando el diagnóstico del análisis inmunofenotípico en biopsia de piel. Las técnicas de NGS, mediante paneles que contengan al menos toda la secuencia codificante de los genes conocidos, permiten obtener un mayor número de casos diagnosticados en una enfermedad heterogénea. Este abordaje se valida en cohortes de casos de EB con mutaciones ya identificadas y también debe ser complementado, ocasionalmente, con estudios específicos para detectar deleciones e inserciones de tamaño intermedio. Aun así, y aunque este abordaje puede aumentar la proporción de pacientes caracterizados en enfermedades genéticas, la NGS no llegará a identificar la mutación en todos los casos, como ocurre en otras enfermedades genéticas. Para dar una solución a los casos en los que no se llega a encontrar la mutación patogénica mediante la secuenciación directa del gen, se emplean técnicas complementarias para la búsqueda de grandes deleciones y duplicaciones, como es el análisis por multiplex ligation probe amplification o arrayCGH. La incorporación de la mutación en la clasificación final de la EB será crítica para proporcionar un asesoramiento genético riguroso, ya que determina el modo de transmisión genética y permite realizar también un pronóstico teórico de la evolución de la enfermedad en cada caso. La realización del pronóstico según hallazgos genéticos debe hacerse con cautela, ya que la influencia de factores ambientales o modificadores de los factores genéticos puede ser determinante en la evolución de la enfermedad2.
Como ya hemos mencionado en varias ocasiones, los genes involucrados en la EB son muchos y, además, las posibles mutaciones en cada uno de ellos son también abundantes15; por tanto, se trata de una enfermedad con heterogeneidad genética alélica (muchas posibles mutaciones) y de loci (diversos genes). Se han identificado mutaciones de todo tipo implicadas en la enfermedad de la EB, desde codón de parada prematura a cambio en la pauta de lectura, entre otras. También su localización es diversa, lo que afecta tanto a la secuencia codificante como a las regiones promotoras e, incluso, a regiones intrónicas. A la hora de realizar una correcta anotación e interpretación del potencial patogénico de una mutación presente en un paciente, se deben realizar búsquedas en bases de datos genéticas generales, como la HGMD (HGMD® Human Gene Mutation Database Professional; http://www.biobase-international.com/product/hgmd), que recoge todas las mutaciones publicadas como causantes de enfermedades genéticas en humanos. Existen, además, otras bases de datos que son más específicas, como la de mutaciones en el gen del COL7A1 asociada al Registro Internacional de Pacientes con EBD31 (http://www.deb-central.org/molgenis.do) o la de filamentos intermedios32 (http://www.interfil.org). En caso de que la mutación no esté reportada en las bases de datos generales ni en las específicas, ni tampoco recogida como un polimorfismo en la base de datos de pequeñas variaciones genéticas (Short Genetic Variation database DbSNP; http://www.ncbi.nlm.nih.gov/projects/SNP/Variations_Survey.html), debe descartarse su presencia en un mínimo de 100 individuos controles de una población con el mismo origen étnico. Aunque no es siempre posible, es recomendable la constatación del efecto patogénico de la mutación a nivel de ARN mensajero o de la proteína.
Partiendo de la premisa de que existen descritas más de 1.000 mutaciones en al menos 19 genes, es muy importante contar con toda la información disponible para optimizar la búsqueda y hacer los cálculos de riesgo. El número de mutaciones nuevas que se identifican sigue aumentando año tras año: en el caso de algunos de los genes, como por ejemplo el COL7A1, en su mayoría son específicas de cada familia. Otros datos para tener muy en cuenta son la frecuencia de mutaciones descritas según lugar geográfico o en relación con determinado origen étnico. Siguiendo con el ejemplo del gen del COL7A1, se ha realizado un estudio poblacional en el cual se ha observado que la mutación patogénica c.6527insC está presente en el 46,3% de los alelos de los pacientes españoles con EBDR. Estamos hablando de una mutación muy frecuente en España, que proviene de un único ancestro común, la cual, actualmente, se analiza como primer cribado para el estudio de pacientes con EBD23,33-35. Esta misma mutación se ha encontrado también con alta recurrencia en Chile36, hipotéticamente, como consecuencia de las oleadas de migración española hacia Latinoamérica en diferentes períodos históricos. Otro dato muy valioso es que esta mutación es, además, más prevalente en el sur-oeste de la península (Extremadura y Andalucía). Existen otras mutaciones frecuentes en España y, aunque su prevalencia es muchísimo menor, su cribado está incluido también en el estudio de portadores que se realiza a las parejas tanto de los propios pacientes como de familiares portadores de una mutación en el gen del COL7A1. El hecho de no portar estas mutaciones recurrentes, aunque no descarta que el probandus tenga otra mutación en alguna de las regiones de gen del COL7A1 no analizadas, reduce drásticamente el riesgo poblacional.
En España no han existido centros de referencia para la EB47 hasta el presente año en el que el Hospital Universitario de La Paz en Madrid y el Hospital Clínic i Provincial de Barcelona y Hospital de Sant Joan de Déu en Cataluña han obtenido dicha designación. En este contexto, el grupo de investigación de la Prof. Marcela del Río (termeg.uc3m.es) realiza desde el 2006 el diagnóstico genético de los pacientes con EB (ORPHA361523; ORPHA361536, ORPHA362075; ORPHA361538), está implicado en proyectos de investigación encaminados al desarrollo de tratamientos para la EBDR (TRA-049, CELLCAM, ORPHA300805, GENEGRAFT; ORPHA437567; ORPHA437569) y en la elaboración del registro nacional de EB (https://registroraras.isciii.es). Este grupo, adscrito al CIBER de Enfermedades Raras (www.ciberer.es), forma parte de la red internacional EB CLINET-Clinical Network of EB Centres and Experts (http://www.eb-haus.org/en/eb-clinet.html;http://www.eb-clinet.org/maps/eb-clinical-contacts.html). El grupo realiza el estudio genético en colaboración con los servicios de dermatología y de genética de diversos hospitales distribuidos por todo el territorio nacional, especialmente con la división de Dermatología Pediátrica del Hospital La Paz (Dr. Raúl de Lucas), recientemente acreditado como centro de referencia y con el Departamento de Genética de la Fundación Jiménez Díaz (Dra. María José Trujillo-Tiebas; Dra. Carmen Ayuso), en sintonía con la asociación de pacientes con EB, DEBRA España (www.pieldemariposa.es; info@debra.es). Esta actividad cooperativa a nivel nacional e internacional ha permitido al grupo alcanzar un conocimiento profundo de la enfermedad8,19,21-26,33,35,37-51. De este modo, frente a la sospecha de una EB, además de solicitar información a DEBRA-España, es conveniente ponerse en contacto con la Dra. María José Escámez (mj.escamez@ciemat.es), coordinadora de los estudios moleculares llevados a cabo por el grupo, para abordar correctamente el estudio genético del paciente.
El asesoramiento o consejo genéticoUna vez identificada la mutación causal, verificar el patrón hereditario es fundamental para establecer en términos probabilísticos el riesgo de recurrencia en futuros hermanos e hijos.
Epidermólisis bullosa autosómica dominanteHasta el momento, se han descrito formas dominantes en la EBS y en la EBD. Las formas dominantes son mayoritarias en la EBS mientras que en EBD tan solo representan, por ejemplo en la cohorte española, entre el 7 y el 8% de los casos23 (126 familias estudiadas). Como en cualquier enfermedad genética con herencia autosómica dominante, basta con portar la mutación en una de las copias del gen para padecer la enfermedad. En estos casos, la persona afectada puede transmitir la mutación (y la enfermedad) a su descendencia con una probabilidad del 50% en cada gestación. Generalmente, en la EBDD suele verse que la persona afectada ha heredado su enfermedad de uno de sus progenitores, que sería también afectado (mutación dominante heredada). Sin embargo, en la EBS dominante, en muchos casos, el paciente es el primer caso de la familia debido a una mutación de novo en línea germinal en progenitores no afectos (10 familias con EBS con mutaciones en KRT5 y KRT10 de novo de un total de 23 familias con mutaciones en estos genes en las que se ha podido confirmar la herencia). Esto se debe a que los genes que codifican las queratinas K5 y K14 presentan sitios bien conocidos de alta inestabilidad genética («hot spot») que son más susceptibles de mutar en los gametos de los progenitores. Este fenómeno ocurriría también en el gen que codifica el Col VII, aunque con menos frecuencia en la cohorte española (2 familias con EBDD de novo de 126 familias de EBD estudiadas). En cuanto al riesgo de recurrencia en futuros hermanos del probandus, este no es del 50%, como ocurre en mutaciones heredadas, sino que se estima en aproximadamente 2-5%52, debido a la posibilidad de que alguno de los progenitores tenga la mutación en mosaicismo en la línea germinal. Cabe resaltar que el riesgo de recurrencia en hijos del probandus sí es del 50%. En casos de pacientes con formas dominantes de novo, el asesoramiento a cargo de un genetista clínico experto se hace indispensable.
Aprovechamos, también, para resaltar que existe una tendencia en los clínicos a hacer un diagnóstico genético de sospecha de herencia dominante de novo en pacientes con formas distróficas leves y padres no afectos. Dada la mencionada baja frecuencia de la aparición de mutaciones dominantes de novo en el gen del Col VII, este tipo de diagnóstico debería evitarse hasta tener el resultado genético y la forma de herencia confirmada. Esto es debido a que, en la mayoría de los casos, este tipo de pacientes presentan en realidad mutaciones recesivas que resultan en presentación clínica leve.
Epidermólisis bullosa autosómica recesivaComo se mencionó anteriormente, la mayor parte de las veces en que vemos un caso de EBD que aparece por primera vez en una familia debemos pensar en una herencia autosómica recesiva, en la que, para producirse la enfermedad, es necesario que ambas copias del gen (paterna y materna) estén mutadas. En estos casos, los padres son portadores sanos (heterocigotos) de una de las 2mutaciones presentes en el ADN de su hijo enfermo. Y el riesgo familiar de que aparezcan nuevos casos afectos es del 25% para los siguientes hermanos de los pacientes. En estos casos es conveniente que los hermanos sanos de afectos, cuando alcancen la mayoría de edad, se estudien también si quieren saber si son portadores o no. Es importante señalar que un portador solo podría tener hijos afectos si su pareja es igualmente portadora. La probabilidad de que, por azar, la pareja sea portadora es menor de uno de cada 50 (2%), excepto si la pareja es consanguínea (o de un área geográfica cercana o con alta prevalencia de una mutación asociada a la EB). Una vez que se identifica la mutación en un portador, cabe la posibilidad de rastrear ese gen en la pareja para descartar razonablemente mutaciones y, por tanto, descartar su estatus de portador.
Esta información respecto del patrón hereditario, y sus implicaciones reproductivas, incluyendo las opciones descritas más abajo de diagnóstico prenatal y preimplantatorio, entre otras, se proporciona en una consulta llamada consejo o asesoramiento genético. Este proceso está recogido en nuestro ordenamiento jurídico (Ley de Investigación Biomédica LIB 14/2007 y la Orden SSI/1356/2015, de 2 de julio de cartera de servicios del SNS) que lo define como el procedimiento destinado a informar a una persona sobre las posibles consecuencias para él o su descendencia de los resultados de un análisis o cribado genéticos y sus ventajas y riesgos y, en su caso, para asesorarla en relación con las posibles alternativas derivadas del análisis. Tiene lugar tanto antes como después de una prueba o cribados genéticos e incluso en ausencia de los mismos. Debe proporcionarse de manera no directiva y cumpliendo los principios de beneficencia y no maleficencia que permitan al paciente tomar sus propias decisiones.
Este modo de proceder es común para cualquier enfermedad genética o hereditaria, y se lleva a cabo de igual manera en EB. Se han publicado algunas recomendaciones sobre el asesoramiento de los pacientes de EB y sus familias52,53, aunque la aplicación de nuevas técnicas hace necesario revisar su disponibilidad y alcance.
Diagnóstico prenatalUna vez detectada la mutación en la persona afecta por la EB, el diagnóstico prenatal es técnicamente abordable54. Para ello, la pareja es atendida en una consulta de genética, donde recibe asesoramiento o consejo genético. La pareja recibe información sobre el tipo de opciones reproductivas a las que puede optar, así como sobre las técnicas obstétricas que se utilizan para acceder al ADN del feto (a partir de células presentes en líquido amniótico o en vellosidades coriales), las posibilidades diagnósticas y los riesgos o complicaciones de estas técnicas, ya que son invasivas. Solo se puede realizar el diagnóstico prenatal si se conoce la mutación y el gen responsable de la EB en esa familia concreta.
Además del estudio directo de la mutación familiar, en algunos tipos de EB, otras técnicas permiten apoyar o realizar el diagnóstico prenatal en casos muy concretos. Por ejemplo, la detección de un aumento de α-fetoproteína en líquido amniótico junto con polihidramnios durante el embarazo justifica la sospecha de una forma específica de EB que cursa con atresia de píloro (EB-PA), detectable por ecografía durante el periodo fetal55, que resulta en muchos casos ser letal. La EB-PA se debe a defectos en las integrinas α6β4 o en la plectina22,40. Por otro lado, en el primer trimestre, las vellosidades coriónicas expresan claramente ambas proteínas y su expresión persiste durante todo el embarazo. De modo que el análisis de las vellosidades coriónicas por inmunofluorescencia representa una herramienta para el diagnóstico prenatal de la EB-PA56. Desafortunadamente, dado que el aumento de la α-fetoproteína y el polihidramnios son síntomas inespecíficos del embarazo que pueden estar asociados a otras enfermedades y la baja casuística de la EB-PA, el obstetra difícilmente los relaciona con esta enfermedad de fragilidad cutánea, si no hay antecedentes familiares.
Una de las novedades en este campo es el diagnóstico prenatal no invasivo que se basa en el estudio del ADN fetal que se encuentra presente en plasma materno. La gran ventaja frente al diagnóstico prenatal convencional es que, al realizarse a partir de una muestra de sangre materna, no implica ningún riesgo para el feto ni para la madre. El diagnóstico prenatal no invasivo de enfermedades monogénicas se hace únicamente en el ámbito de la investigación y para algunas mutaciones. En la muestra de plasma materno, el ADN fetal coexiste con cantidades mucho mayores de ADN de origen materno. La coexistencia de los 2ADN en la muestra limita estos estudios al análisis de regiones que hayan sido heredadas de novo o vía paterna y que no estén presentes en el genoma materno, para poder asegurar su origen fetal en caso de ser detectadas en la muestra. A pesar de esta limitación, se ha demostrado la eficacia de este método para la detección de mutaciones57.
Diagnóstico preimplantacionalHoy en día, además, existe la posibilidad, mediante técnicas de reproducción asistida, de realizar el diagnóstico de embriones y la posterior implantación en el útero materno de aquellos que se encuentren libres de la enfermedad. Es lo que se conoce como diagnóstico genético de preimplantación (DGP). El DGP, disponible en España tras la aprobación de la Ley 14/2006, de 26 de mayo, sobre técnicas de reproducción humana asistida, ofrece una opción reproductiva alternativa a las familias que presentan enfermedades hereditarias graves, de aparición precoz y para las que todavía no existe tratamiento curativo, entre las que se encuentra la EB. Se recomienda que las parejas recurran a un Servicio de Reproducción Asistida con experiencia en el manejo de este tipo de diagnóstico, que trabaje coordinadamente con un Servicio de Genética y que tenga optimizados los protocolos de fertilización in vitro, generalmente extensos, con el objeto de conseguir un número adecuado de embriones para analizar.
La primera premisa para poder realizar el DGP es disponer del diagnóstico genético preciso de la pareja, es decir, que se hayan identificado las mutaciones concretas que portan ambos progenitores. Es necesario, además, realizar un estudio de «informatividad» en la pareja, para elegir los marcadores genéticos más adecuados. Es decir, el estudio de marcadores genéticos que acompañan a la mutación y que permiten diferenciar el haplotipo que se asocia a la enfermedad. Estos marcadores se analizan en esa pareja en particular para estimar las posibles combinaciones en los posibles futuros embriones que se generen. De este modo podremos saber, a priori, si la combinación de marcadores de ambos progenitores permitirá distinguir, sin lugar a dudas, los embriones afectos de los sanos. Si el estudio de marcadores genéticos resultase «no informativo», esto es, que las combinaciones de marcadores hacen indistinguibles los embriones sanos de los afectos, el diagnóstico genético de preimplantación podría no ser factible técnicamente, dependiendo de la estrategia diagnóstica posterior.
En el caso de que la pareja resultase apta para el estudio, ha de seguirse un protocolo similar al de una fecundación in vitro con inyección intracitoplásmica de espermatozoides convencional, si bien en el caso del DGP es necesario conseguir un número más alto de óvulos. Así, la futura madre ha de seguir un tratamiento farmacológico de estimulación ovárica controlada, consistente en la inyección de hormonas estimulantes con la finalidad de que maduren varios óvulos en el mismo ciclo. Tras la extracción de los óvulos (folículos maduros) se fecundan in vitro para obtener los embriones. A los 3días de la fecundación, cuando los embriones están formados por 6-8 células (inicio de la mórula), se biopsia una célula (blastómero) de cada uno y se envía al laboratorio de genética para realizar el diagnóstico que dictaminará qué embriones son portadores de la enfermedad y cuáles no (embriones sanos). Este estudio debe realizarse en un período no superior a 24 h.
De los embriones considerados sanos respecto de la mutación y, por tanto, transferibles, se han de seleccionar de nuevo desde el punto de vista embriológico aquellos embriones (nunca más de 3) que presentan mejores características morfológicas para ser transferidos al útero materno y que proporcionen mayor garantía de éxito para su implantación. Al transferirse varios embriones no es infrecuente que se implante más de uno y que se desarrolle un embarazo múltiple. Si fracasa el proceso por alguna razón, la mujer puede iniciar un nuevo ciclo de estimulación ovárica para comenzar de nuevo el proceso.
Durante el proceso de consentimiento informado, se debe informar a la pareja de sus resultados particulares esperables, y de las tasas de éxito y de fracaso y, posteriormente, de los resultados que se vayan obteniendo a lo largo de todo el proceso. Por ejemplo, deben conocer que la tasa de embarazo de estas técnicas es bajo (menos del 20% en cada ciclo que se intente). El DGP tiene una fiabilidad comprendida entre el 88 y el 96%58, por lo que se recomienda un diagnóstico prenatal convencional una vez instaurado el embarazo para confirmar los resultados.
En España, el diagnóstico genético prenatal y el de preimplantación están incluidos dentro de las prestaciones del SNS, cuando se cumplen los criterios establecidos de riesgo genético, características de la descendencia y estudio de fertilidad y edad de la pareja. Por tanto, el procedimiento es gratuito para las parejas portadoras de EB que cumplan estos criterios, a través del sistema sanitario público. Cada comunidad autónoma dispone de centros de referencia para el estudio genético preimplantacional. Por ello, las parejas deben consultar en su respectiva comunidad autónoma. Este proceso está recogido en la cartera de servicios del SNS, que es válida para todo el estado y está regulado por la Ley de Reproducción Humana Asistida y por la Comisión de Reproducción Humana Asistida para los casos que planteen alguna situación no contemplada por la ley.
FinanciaciónEl grupo de la Dra. del Río y, en particular, el diagnóstico molecular han sido financiados por fondos específicos del CIEMAT, el IIS-FJD, los proyectos CELLCAM, GENEGRAFT HEALTH-F2-2011-261392, SAF2013-43475-R, ICI14/00363, CIBERER INTRAMURAL/08/714.1, por la Fundación Ramón Areces (CIVP-16A1864) y por parte de las Asociaciones de Pacientes Berritxuak y, más recientemente, por DEBRA-España. El grupo de la Dra. Ayuso agradece los recursos obtenidos por el PIE13/00051, CIBERER CB06/07/0036 y Biobanco PT13/0010/0012.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Queremos agradecer a las personas con EB y sus familias la confianza, el respeto y la voluntad de colaborar con nuestro equipo que siempre han mostrado.
A la Asociación Piel de Mariposa DEBRA-España.
A los profesionales sanitarios de enfermería, genetistas, dermatólogos, pediatras, cirujanos y de otras especialidades que, de alguna manera, han participado en la realización del diagnóstico molecular, sin cuya colaboración y dedicación este trabajo no hubiera sido posible. En especial, a nuestros colaboradores más asiduos por dedicarnos un tiempo del que carecen, ponernos en contacto con sus pacientes, facilitarnos la obtención de muestras e información (incluidas algunas de las fotografías clínicas que ilustran este trabajo). No queremos olvidarnos tampoco de los grupos del Dr. Meneguzzi (INSERM-Francia) y la Dra. Zambruno (Istituto Dermopatico dell’Immacolata, actualmente, Ospedale Pediatrico Bambino Gesù-Italia), líderes mundiales en el ámbito de la EB, por su ayuda en nuestros inicios.
Al equipo de diagnóstico de EB, Nuria Illera, Almudena Holguín, Dra. Adela García, Dra. Marta García, Dr. Álvaro Meana, Dra. Sara Llames, Eva García y Dra. Ángeles Mencía por el excelente trabajo que realizan y por su dedicación.
La colaboración entre los grupos de la Dra. Carmen Ayuso y la Dra. Marcela del Río ha sido fundamental en el traslado del diagnóstico molecular al paciente mediante el asesoramiento genético y el diagnóstico prenatal.
Cátedra Fundación Jiménez Díaz de Medicina Regenerativa y Bioingeniería Tisular: http://www.fjd.es/iis_fjd/es/areas-grupos-investigacion/tecnologia-innovacion-sanitaria/grupos-asociados/medicina-regenerativa-ingenieria-tejidos.
Asociación de Epidermólisis Bullosa en España. www.pieldemariposa.es/
Profesionales sanitarios: Dr. Raúl de Lucas, Dra. Rocío Maseda y Dr. Juan Carlos López (Hospital La Paz, Madrid), Dra. Ángela Hernández y Dr. Antonio Torrelo (Hospital Niño Jesús, Madrid), Dr. José Lasso (Hospital Universitario Gregorio Marañón, Madrid), Dra. Asunción Vicente, Dra. Loreto Martorell (Hospital St Joan de Deu, Barcelona), Dra. Pilar Iranzo, Dra. Susana Puig y Dr. José Manuel Mascaró (Hospital Clínic, Barcelona), Dra. Raquel Rodríguez y Dra. Isabel Febrer (Consorcio Hospital General, Valencia), Dr. Agustí Toll, Dr. Ramón Pujol (Hospital del Mar, Barcelona), Dra. Yolanda Gilaberte (Hospital San Jorge, Huesca), Dr. Juan Luis Santiago (Hospital Ciudad Real), Dr. José Bernabé (Hospital Virgen del Rocío, Sevilla), Dr. Ángel Vera (Hospital Carlos Haya, Málaga), Dra. Eulalia Baselga (Hospital Santa Creu i Sant Pau, Barcelona) y Dres. Sebastián Mir-Mir y Roger Costa (Hospital Plató de Barcelona), Dra. Raquel Sáez (Hospital Universitario de Donostia, San Sebastián), Dra. Elena Arana (Hospital Vall d’Hebron, Barcelona) y Dr. José Carlos Moreno (Hospital Reina Sofía, Córdoba).


