







Las dermatosis purpúricas pigmentadas son un grupo de enfermedades benignas y de curso crónico. Las variantes descritas representan distintas formas clínicas de una misma entidad con unas características histopatológicas comunes para todas ellas. Exponemos a continuación un resumen de las variedades más frecuentes, sus características clínicas, dermatopatológicas y de epiluminiscencia. Al tratarse de una entidad clínica poco frecuente, benigna, y no conocerse claramente los mecanismos patogénicos de la misma, no existen tratamientos estandarizados. Se revisan los tratamientos publicados hasta el momento, la mayoría de ellos basados en casos aislados o pequeñas series de casos, sin poder establecer un nivel de evidencia suficiente como para ser recomendado ninguno de ellos como tratamiento de elección.
The pigmented purpuric dermatoses are a group of benign, chronic diseases. The variants described to date represent different clinical presentations of the same entity, all having similar histopathologic characteristics. We provide an overview of the most common PPDs and describe their clinical, dermatopathologic, and epiluminescence features. PPDs are both rare and benign, and this, together with an as yet poor understanding of the pathogenic mechanisms involved, means that no standardized treatments exist. We review the treatments described to date. However, because most of the descriptions are based on isolated cases or small series, there is insufficient evidence to support the use of any of these treatments as first-line therapy.
Las dermatosis purpúricas pigmentadas (DPP) son un grupo de enfermedades poco frecuentes, benignas y de curso crónico, que se caracterizan por la aparición de múltiples petequias sobre máculas hiperpigmentadas pardo-amarillentas1. Todas ellas representan distintas formas clínicas de una misma entidad, con unas características histopatológicas comunes2,3.
Clásicamente se han descrito 5 variantes: enfermedad de Schamberg o DPP progresiva, púrpura pruriginosa o púrpura eccematoide de Doucas y Kapetanakis, DPP liquenoide de Gougerot y Blum, liquen aureus o purpúrico y púrpura anular telangiectoide o enfermedad de Majocchi1. Además, podemos encontrar otras formas clínicas menos frecuentes como la DPP granulomatosa, la púrpura pruriginosa de Loewenthal, la DPP lineal, la DPP transitoria y las formas familiares2.
EpidemiologíaLas DPP son trastornos infrecuentes, siendo la enfermedad de Schamberg la forma más habitual de presentación1. Los adultos son los principales afectados, aunque también se han descrito casos en niños4, en los que la enfermedad de Schamberg es también la presentación más común5. En general las DPP son más frecuentes en hombres1, especialmente en su forma lineal6, mientras que la enfermedad de Majocchi es más frecuente en mujeres5.
EtiopatogeniaAunque su etiología es desconocida, distintos factores como el ejercicio físico, la hipertensión venosa, la diabetes mellitus, o las infecciones2,7 se han relacionado con su aparición. También se han asociado con el consumo de distintos fármacos (tabla 1)8, y en la variante granulomatosa se ha objetivado una relación con dislipemia y trastornos autoinmunes9. Sin embargo, en la mayoría de los casos no se llega a detectar un agente causal10.
Fármacos relacionados con las DPP
| Sedantes/hipnóticos | Fenobarbital, clordiazepoxido, meprobamato |
| Vitaminas | Tiamina (B1) |
| Diuréticos | Furosemida |
| Cardiovascular | Nitroglicerina, bezafibrato, hidralazina, dipiridamol, sildenafilo |
| Antibióticos | Ampicilina |
| Analgésicos | AINE, aspirina, acetaminofeno |
| Estimulantes | Pseudoefedrina |
| Hormonas | Acetato de medroxiprogesterona |
| Antidiabéticos | Glipizida |
| Quimioterápicos | 5-Fluouracilo tópico |
| Antivirales | Interferón alfa |
| Retinoides | Isotretinoína |
Fuente: Kaplan et al.8.
Respecto a los mecanismos fisiopatológicos propuestos se encuentran la dilatación y la fragilidad capilar10. Se ha hipotetizado que las células responsables son las que conforman la estructura de los vasos, como los fibroblastos y las células endoteliales. Ante ciertas condiciones de activación, por ejemplo, ante una presión intravascular elevada, o bien espontáneamente, estas células pueden alterar su función y producir una fuga de hematíes a través de las paredes vasculares11. Esto desencadenaría una reacción de hipersensibilidad mediada por células. Por lo tanto la respuesta inmune celular tiene un papel fundamental en la patogenia de las DPP12,13. El infiltrado inflamatorio perivascular está conformado por linfocitos CD4+14 (en los que existe una reducción de la expresión de CD715) y células dendríticas CD1a+12.
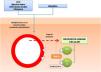
Además, en distintos estudios se ha analizado el papel de las moléculas de adhesión celular en el desarrollo de las DPP. Se trata de proteínas de membrana que interactúan con ligandos específicos que producen y mantienen puentes entre las células o con proteínas de la matriz extracelular. Se ha objetivado una intensa expresión de las moléculas de adhesión LFA-1 (antígeno de función linfocitaria 1) e ICAM-1 (molécula de adhesión intercelular 1) en las células inflamatorias, y de ICAM-1 y ELAM-1 (molécula de adhesión leucocitaria endotelial 1)12 en las células endoteliales. De esta manera los linfocitos T que son activados por un estímulo antigénico se adhieren a las células endoteliales, fibroblastos y queratinocitos16. Citoquinas producidas por leucocitos, como TNF-α, pueden desencadenar la expresión de estas moléculas de adhesión (fig. 1).
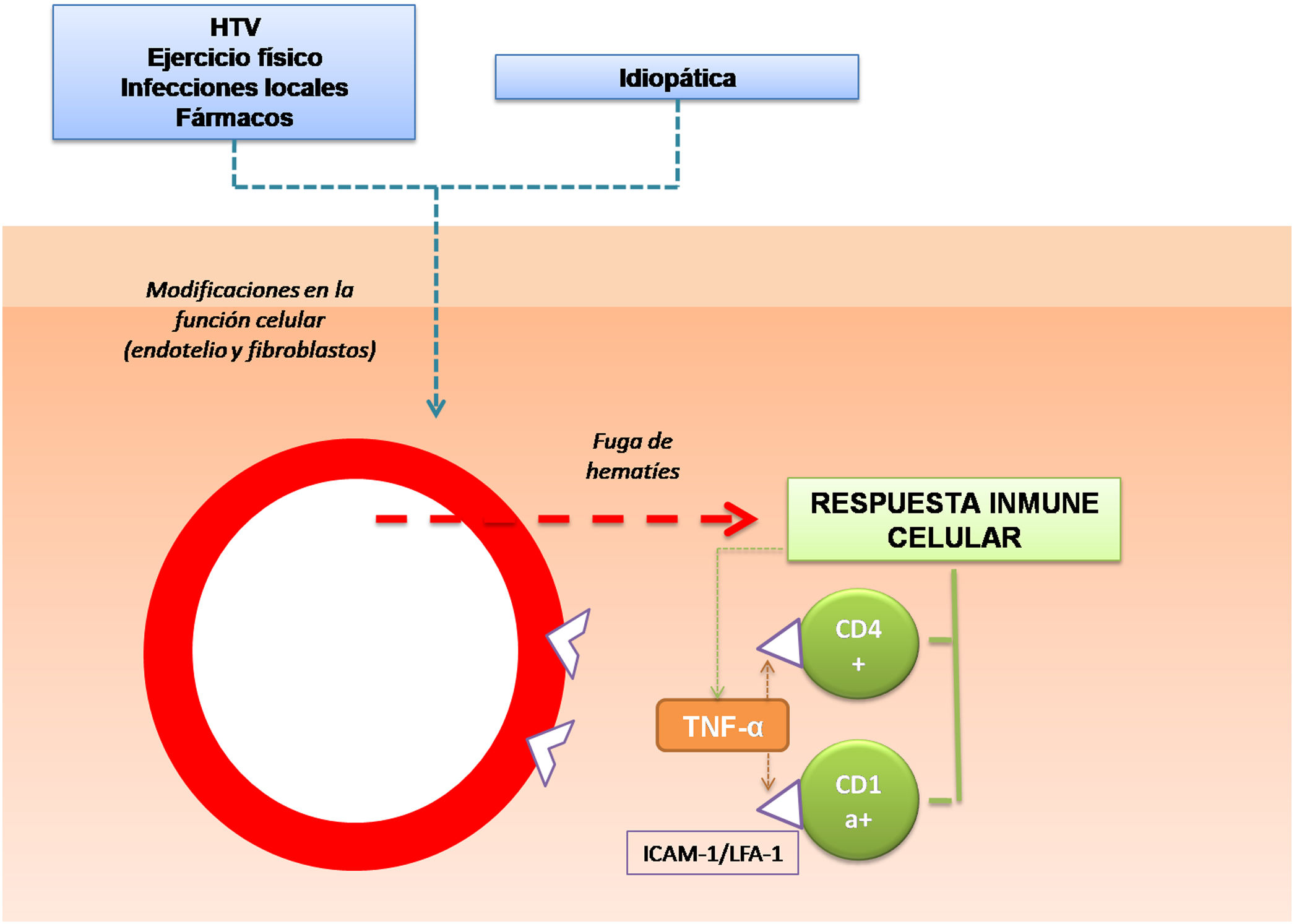
Estas mismas citoquinas podrían provocar una menor liberación del activador del plasminógeno endotelial y/o un aumento excesivo del inhibidor del activador del plasminógeno17, lo que contribuiría a la reducción de actividad fibrinolítica y al depósito intraperivascular de fibrina que aparece en estas patologías18.
Además, en estudios de inmunoflorescencia directa se puede observar depósitos de fibrinógeno, IgM y/o C3 en los vasos superficiales de la dermis10.
Otras hipótesis surgidas en los últimos años sugieren que las DPP representan una alteración insidiosa y epiteliotrópica de linfocitos T. Datos como la detección de epidermotropismo o monoclonalidad en el infiltrado inflamatorio van a favor de ello15,19. Incluso existen algunos casos descritos de progresión a micosis fungoide20–22. La distinción entre una micosis fungoide purpúrica y una variante monoclonal de DPP es compleja y debemos analizar la clínica junto a los datos moleculares e histopatológicos15,20,23. Signos como la aparición de poiquilodermia, prurito o confluencia de las placas, una duración mayor a un año, o la detección de monoclonalidad y una disminución de CD7 y CD62L en el infiltrado debe hacernos sospechar en una progresión, incluso cuando los linfocitos no presentan una atipia importante15,24. Algunos autores se inclinan por tratar como un estadio inicial de micosis fungoide aquellas DPP diseminadas y monoclonales.
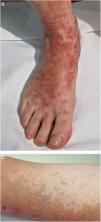
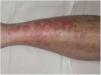
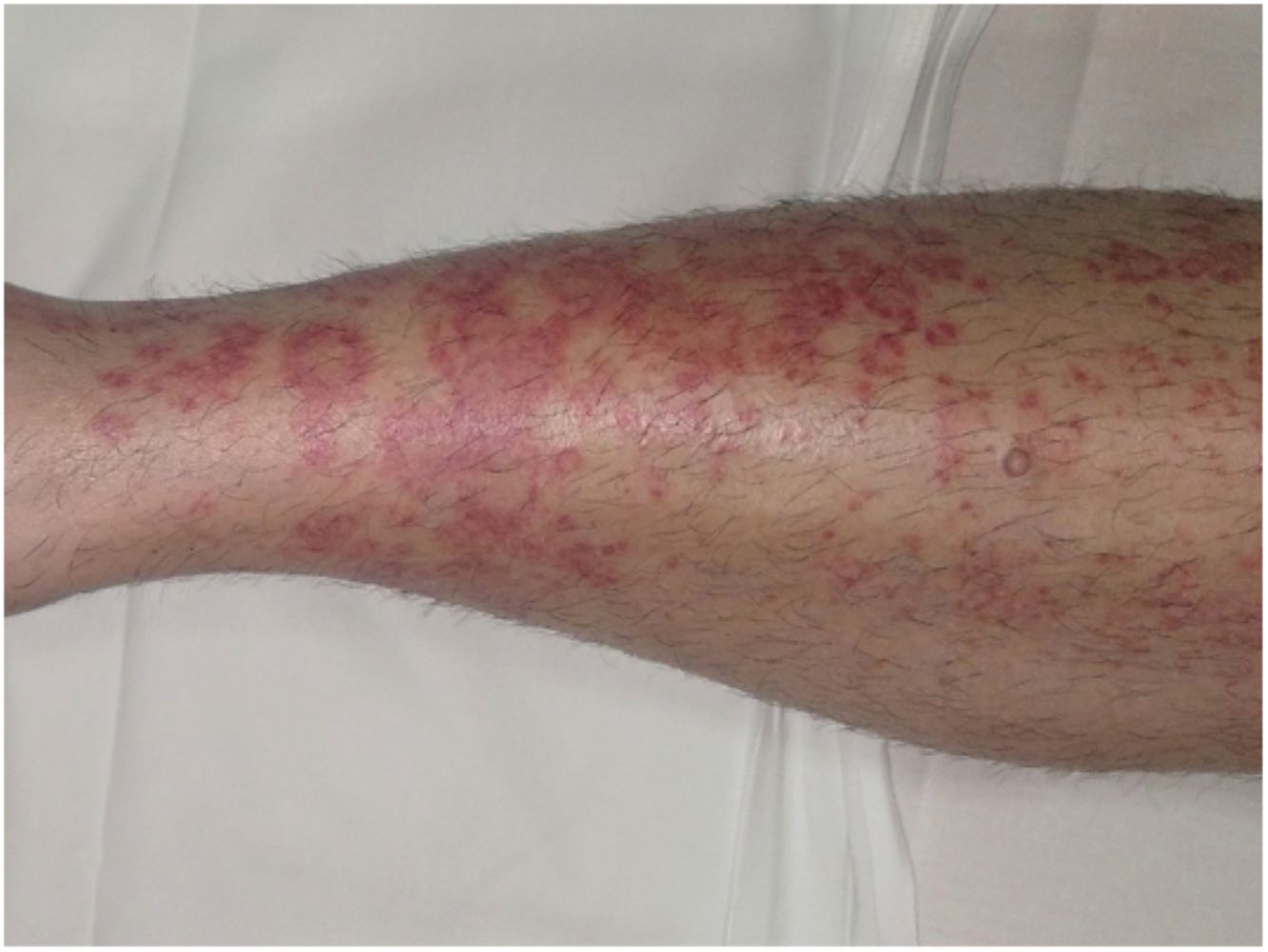
Formas clínicasDermatosis purpúrica pigmentada progresiva o enfermedad de Schamberg1,2Las lesiones suelen aparecer en extremidades inferiores de forma bilateral, pero pueden aparecer en tronco, brazos, muslos y glúteos. Consisten en máculas rojo-anaranjadas que en la periferia presentan un punteado purpúrico en forma de granos de pimienta de cayena (fig. 2a y b). Se tornan en amarillento-parduzcas conforme evolucionan. Son asintomáticas de forma general, aunque hay pacientes que refieren prurito. Su curso es crónico, con numerosas exacerbaciones y remisiones.
Púrpura pruriginosa o púrpura eccematoide de Doucas y Kapetanakis1,25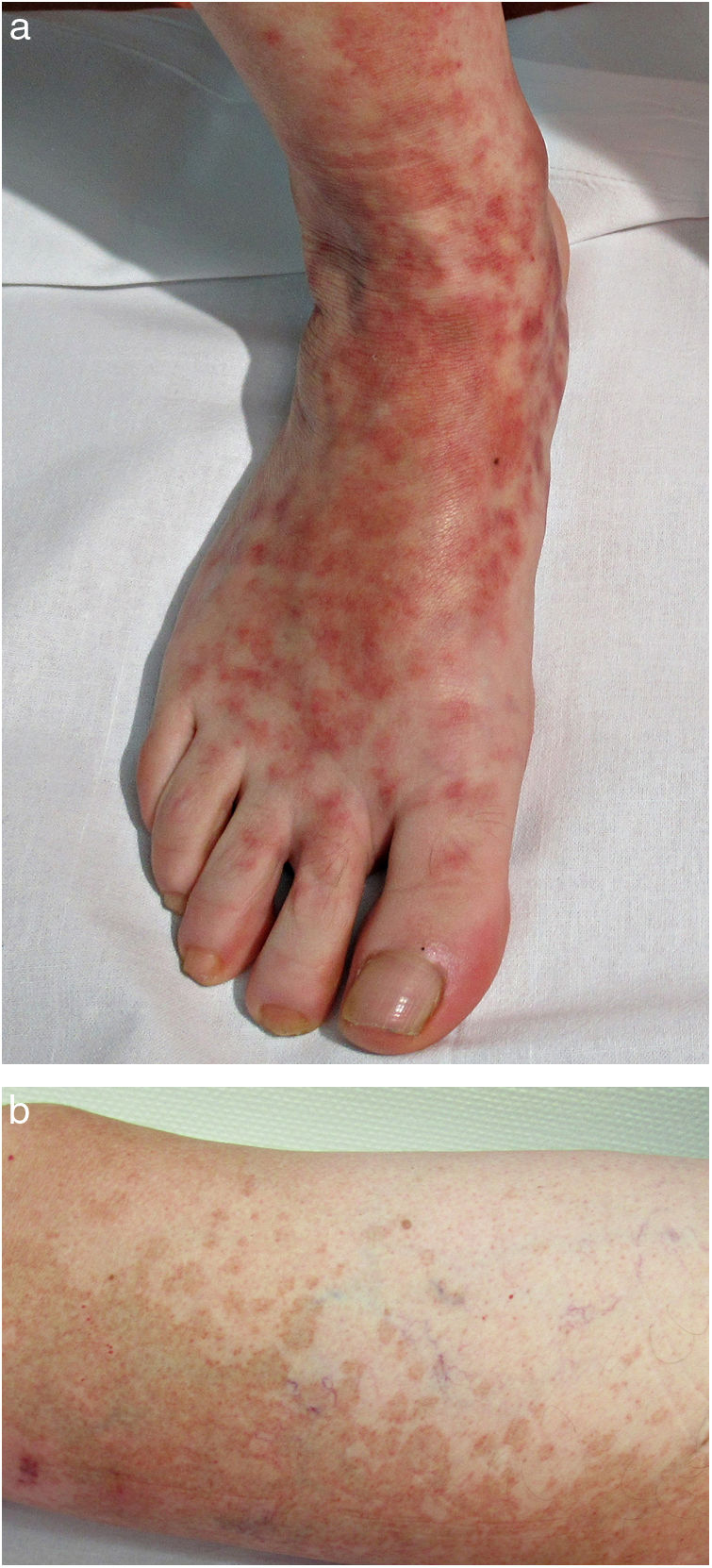
Es la forma más extensa y pruriginosa. La localización más frecuente es las extremidades inferiores, y su presentación clínica es similar a la enfermedad de Schamberg: máculas purpúricas o petequiales, con la peculiaridad de que aparece descamación en su superficie. Se ha asociado a dermatitis alérgica de contacto a caucho y ropa. Su aparición es rápidamente progresiva en 15-30 días, y puede persistir durante meses o años.
Dermatosis purpúrica pigmentada liquenoide de Gougerot y Blum1,2,26Se caracteriza por la aparición de pápulas violáceas liquenoides que tienden a fusionarse formando grandes placas en piernas, aunque en ocasiones puede afectar al tronco. Su curso es crónico y suele afectar a varones de edad avanzada. El diagnóstico diferencial debe realizarse con el sarcoma de Kaposi.
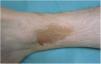
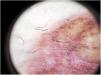
Liquen aureus o liquen purpúrico1,2,27Es una variante más localizada, generalmente única o con escaso número de lesiones, y persistente en el tiempo. Cursa con la aparición brusca de pequeñas pápulas amarillo-anaranjadas, de aspecto liquenoide con tendencia a confluir en placas de entre 1 y 20 centímetros, asociadas a lesiones purpúricas milimétricas (fig. 3). La localización más frecuente son las extremidades inferiores, aunque pueden aparecer en cualquier zona. Suelen ser asintomáticas. En niños y adolescentes se han descrito variantes zosteriformes28 y segmentarias siguiendo líneas de Blaschko29, o la distribución de las venas safena30 y cefálica31.
Púrpura anular telangiectoide o enfermedad de Majocchi1,2,25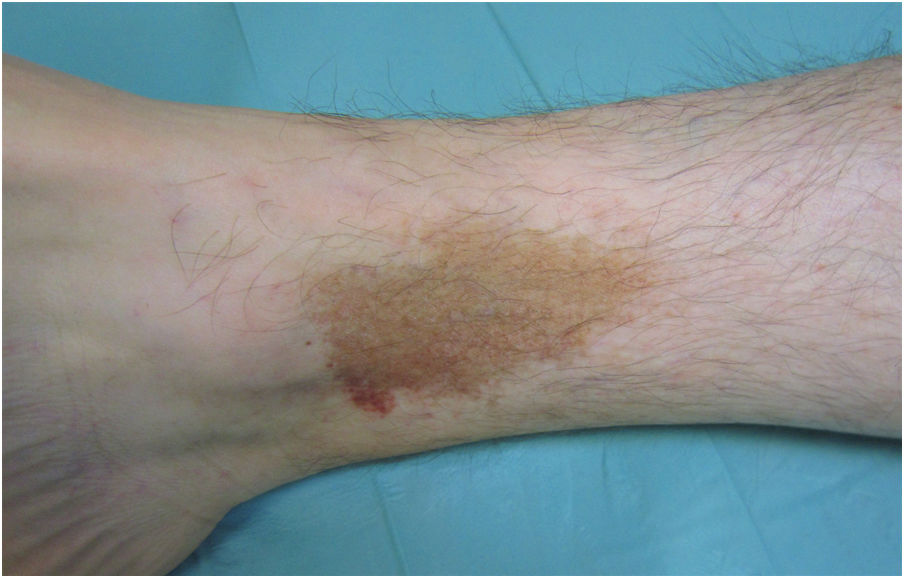
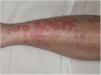
Las lesiones más precoces consisten en máculas rojo-violáceas anulares (fig. 4). Posteriormente aparecen las telangiectasias puntiformes en un tono rojo más oscuro. Crecen periféricamente, mientras que se van aclarando en el centro llegando a producir atrofia. La erupción comienza en las extremidades inferiores y posteriormente se extiende a tronco y brazos alcanzado un gran número de lesiones. Se ha descrito una variante denominada púrpura telangiectásica arciforme32, en la que las lesiones son menos numerosas, pero de mayor tamaño y con una característica morfología arqueada.
Otras variantes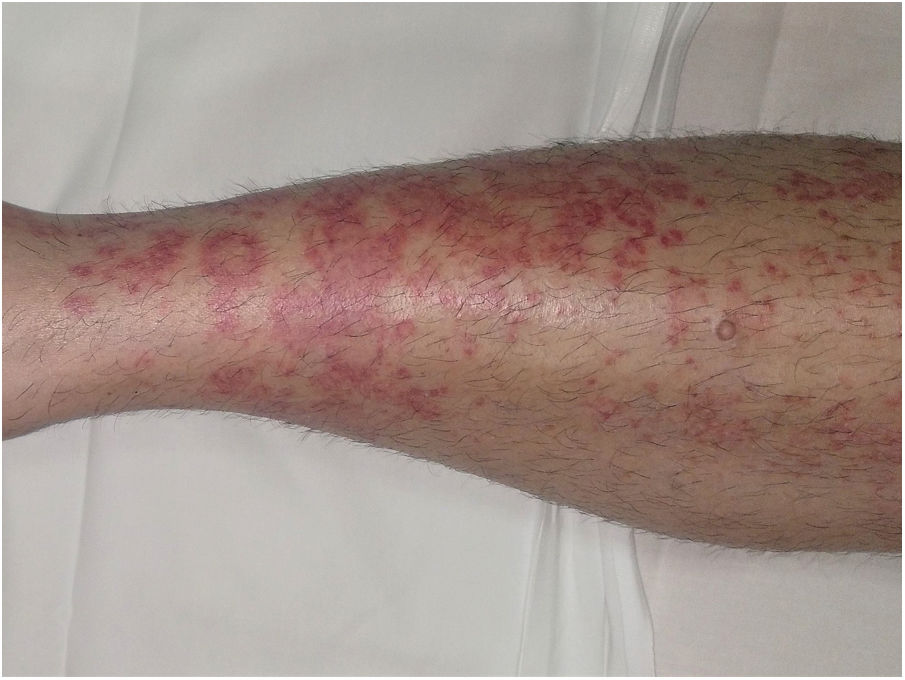
Hersch y Schwayder33 describieron una forma lineal y unilateral que es considerada una rareza y que debe ser diferenciada de las formas lineales de la enfermedad de Schamberg y de liquen aureus. Higgings y Cox34 por su parte describieron una forma cuadrangular cuyo origen era una obstrucción vascular a nivel pélvico.
Existe una variante transitoria35que incluye entidades como el angioma serpiginoso36, un trastorno vascular infrecuente que suele comenzar en la infancia y afecta con más frecuencia al sexo femenino, existiendo evidencias de dependencia estrogénica. Se caracteriza por la aparición de múltiples máculas rojo-purpúricas, asintomáticas, organizadas en pequeños grupos que se distribuyen por las extremidades siguiendo un patrón serpiginoso.
La púrpura pruriginosa de Lowenthal37, tan solo descrita en adultos, es considerada una variante más sintomática de la enfermedad de Schamberg.
La forma granulomatosa descrita por Saito38 es más frecuente en mujeres y se trata de un hallazgo histopatológico indiferenciable a nivel clínico de otras DPP.
Por último, se han reportado formas familiares de enfermedad de Schamberg y de púrpura anular telangiectoide, con patrón de herencia autosómico dominante39.
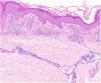
HistopatologíaEste grupo de enfermedades se caracterizan por un infiltrado perivascular linfohistiocitario localizado a nivel de los vasos de pequeño calibre en dermis superficial. Es típico el edema en las células endoteliales y el estrechamiento de la luz vascular10. También es frecuente observar la extravasación de hematíes con depósito de hemosiderina en los macrófagos (fig. 5a y b). Las tinciones de Perls y de Fontana Masson permiten demostrar la presencia de hierro (hemosiderina) en la dermis superficial, lo que diferencia las DPP de la dermatitis de estasis en la que el depósito es más profundo2.
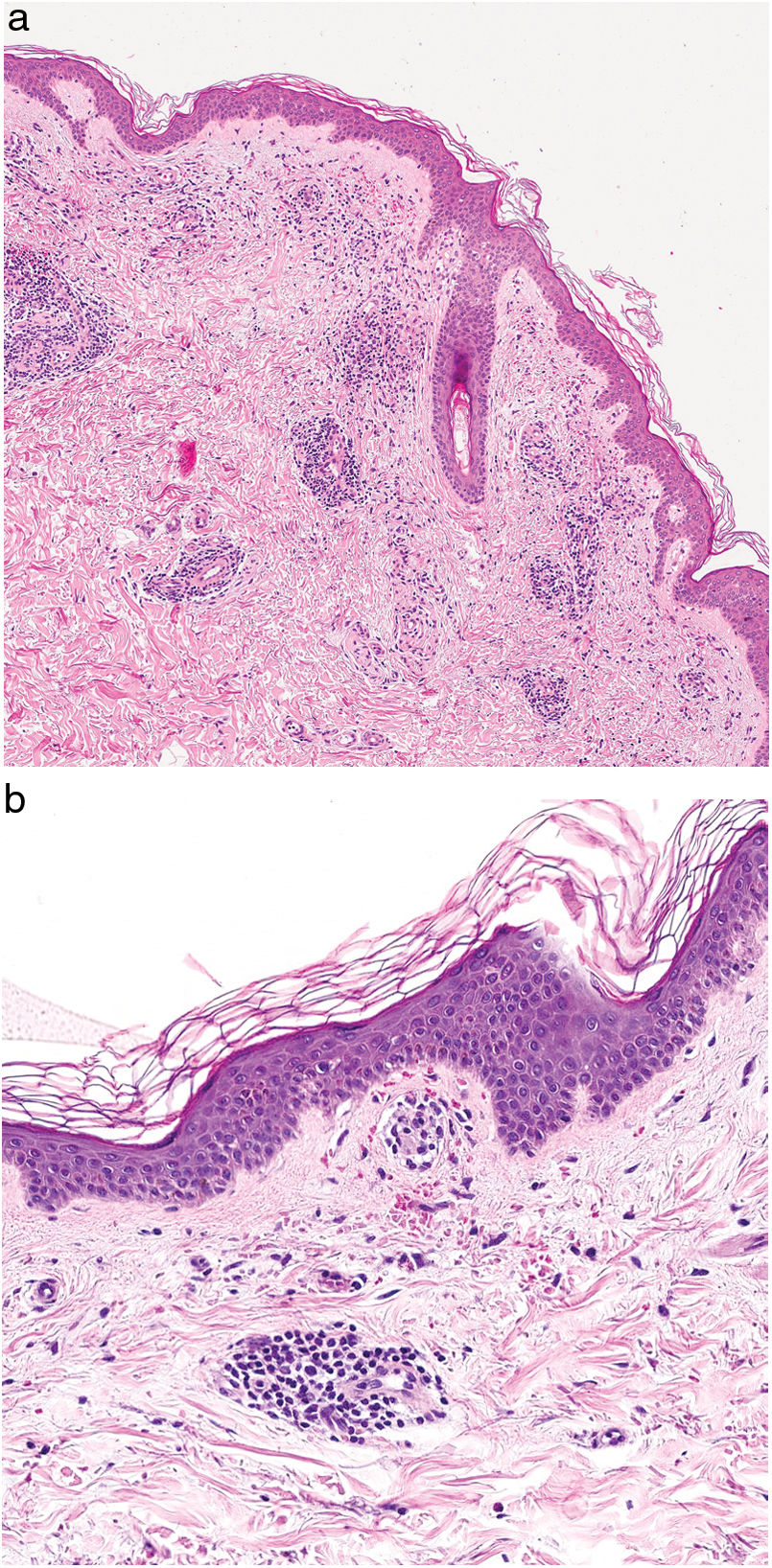
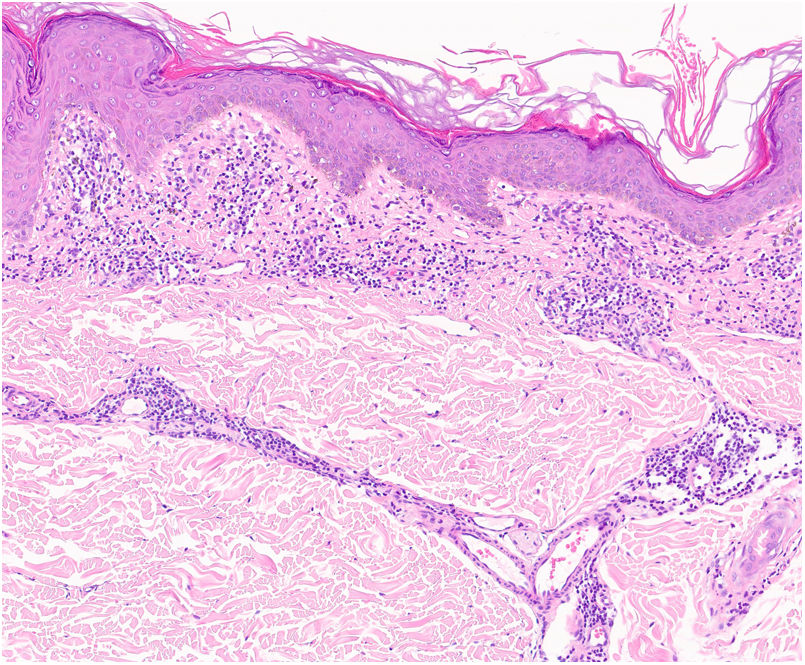
Una epidermis intacta separada de un infiltrado dérmico en banda por una zona de tejido conectivo no afecto (zona de Grenz) caracteriza al liquen aureus27 (fig. 6). Este mismo infiltrado dérmico es característico de las formas liquenoides de Gougerot y Blum26. En cambio, en las variantes eccematoides de Doucas y Kapetanakis destaca la espongiosis epidérmica y la presencia de neutrófilos en el infiltrado25. En las formas granulomatosas a los hallazgos típicos de las DPP se superpone un infiltrado granulomatoso perivascular38. En la tabla 2 se compara la presentación clínica, localización y hallazgos histopatológicos de las variedades más frecuentes.
Características clínicas e histopatológicas de las DPP
| Variantes | Presentación clínica | Localización | Hallazgos histopatológicos |
|---|---|---|---|
| Enfermedad de Schamberg | Máculas rojo-ananjadas con punteado en granos de pimienta de cayena en periferia | Extremidades inferiores En ocasiones: tronco, brazos, muslos y glúteos | Infiltrado linfohistiocitario a nivel de los vasos de pequeño calibre en dermis superficial, hematíes extravasados y depósito de hemosiderina en los macrófagos |
| Púrpura eccematoide de Doucas y Kapetanakis | Similar a la enfermedad de Schamberg, asociando descamación y prurito intenso. | Extremidades inferiores | Infiltrado con mayor cantidad de neutrófilos y espongiosis en la epidermis |
| DPP liquenoide de Gougerot y Blum | Pápulas violáceas liquenoides que confluyen formando placas | Extemidades inferiores | Infiltrado dérmico en banda |
| Liquen aureus | Placa aislada y persistente rojo-anaranjada asociada a lesiones púrpuricas | Extremidades inferiores | Epidermis sin cambios e infiltrado dérmico en banda con zona de Grenz |
| Enfermedad de Majocchi | Placas de crecimiento periférico con telangiectasias puntiformes en los bordes y aclaramiento central | Extremidades inferiores y tronco | Idéntico a la enfermedad de Schamberg |
Mientras que algunos autores1,13 consideran que la capilaritis es el concepto global que podría definir a estas entidades, Ackerman enfatizó en que no lo es, ya que ni se objetiva fibrina en la pared vascular ni trombos en el interior de la luz40.
DiagnósticoAdemás de la biopsia cutánea, se recomienda realizar una analítica sanguínea para descartar trombocitopenia, alteraciones en la coagulación o de la autoinmunidad (anticuerpos antinucleares, factor reumatoide) y la presencia de infecciones crónicas (anti-VHC y anti-HBsAg)2.
Diagnóstico diferencialEl diagnóstico diferencial se establece con entidades que pueden ocasionar cuadros purpúricos a nivel de extremidades inferiores. Recogemos sus principales características en la tabla 3.
Diagnóstico diferencias de las DPP
| Entidades clínicas | Características principales |
|---|---|
| Reacciones de hipersensibilidad a fármacos | Consumo reciente de dichos fármacos |
| Carbamacepina, meprobamato, clordiazepóxido, furosemida, nitroglicerina, vitamina B1 y 5-fluouracilo tópico | |
| Dermatitis de contacto purpúrica por ropa | Lesiones delimitadas inicialmente a las zonas de contacto con la ropa. Prurito intenso |
| Lana, colorantes dispersos | |
| Púrpura por estasis venoso | Signos de insuficiencia venosa crónica: edemas, varices, sensación de pesadez, úlceras venosasDepósito de hemosiderina en dermis profunda |
| Púrpura por trombocitopenia | Asociada a cifras de plaquetas menores a 100-150.000/μl |
| Púrpura senil | En ancianos, que pueden asociar consumo de antiagregantes, anticoagulantes o corticoides |
| Exantema purpúrico secundario a infección viral | Acompañado de sintomatología infecciosa |
| Vasculitis leucocitoclástica | Lesiones purpúricas palpables.HP: necrosis fibrinoide, edema endotelial y leucocitoclastia |
| Púrpura de Schölein-Henoch | De 3 a 15 años. Asocian púrpura simétrica en piernas y glúteos, con dolor articular y abdominal. |
| Sarcoma de Kaposi | En ancianos o inmunodeprimidos. HP: células fusiformes en dermis que configuran luces vasculares irregulares |
| Micosis fungoides purpúricas | Cuadros de >1 año, diseminados, con monoclonalidad en el infiltrado y pérdida de CD7 |
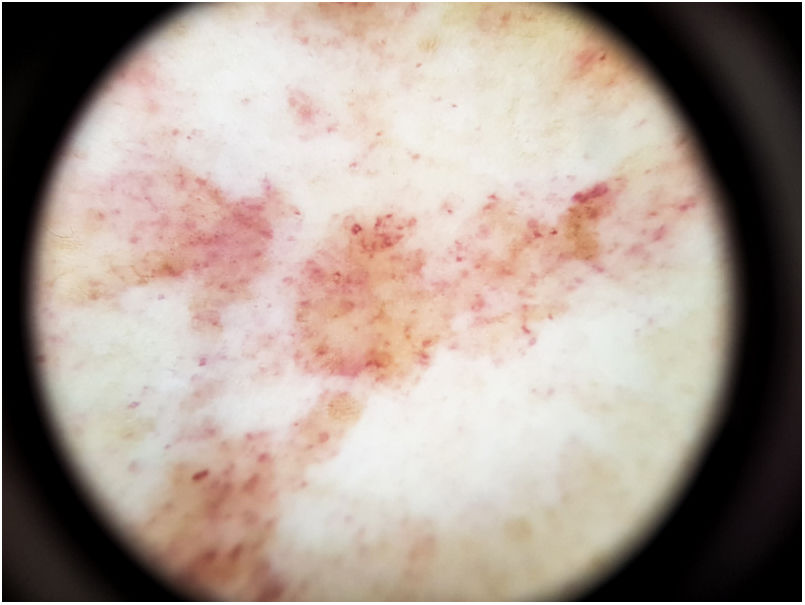
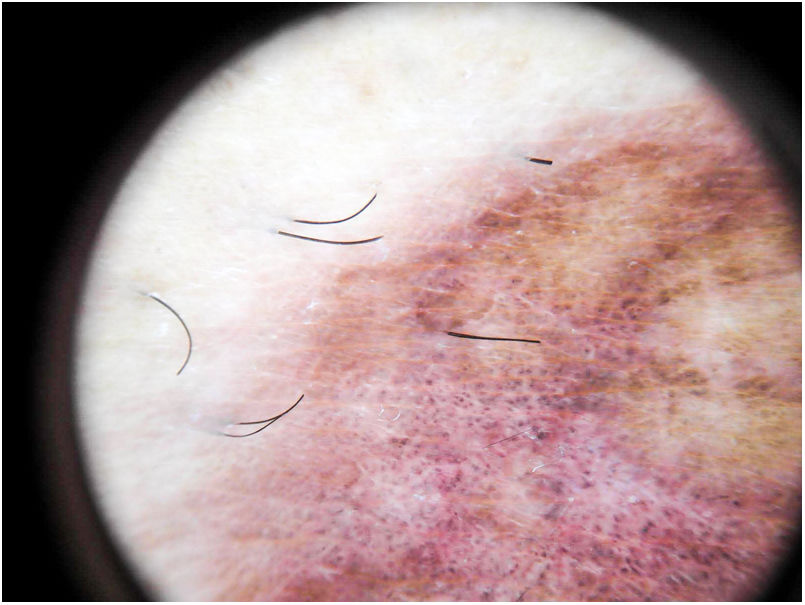
El hallazgo más frecuente es la pigmentación rojo-cobriza difusa en el fondo, que se podría explicar por el infiltrado dérmico linfohistiocitario, la extravasación de hematíes y el depósito de hemosiderina en los histiocitos41. También se observan glóbulos y puntos rojos que se deben a la extravasación de hematíes, y al aumento de número y dilatación de los vasos sanguíneos42 (fig. 7). En casi la mitad de los pacientes se pueden observar puntos marrones, que se explican por la disposición esférica o elíptica de melanocitos y melanófagos en la unión dermoepidérmica. En un tercio de los pacientes se encuentra un pseudoretículo pigmentado que se corresponde con una hiperpigmentación de la capa basal e incontinentia pigmenti en la dermis papilar. Se ha descrito un patrón específico de liquen aureus43 en el que se puede observar un fondo rojo cobrizo, con puntos y glóbulos marrones y rojos, puntos grises y un pseudoretículo formado por líneas pigmentadas interconectadas (fig. 8).
Dada la naturaleza benigna de las DPP y la ausencia de tratamientos estandarizados y con eficacia demostrada, la relación entre el riesgo y el beneficio del tratamiento debe de ser considerada.
Al tratarse de un cuadro benigno y asintomático podríamos no realizar tratamiento alguno4. Pero en muchas ocasiones, la cronicidad, la repercusión física que conlleva el cuadro, la extensión de las lesiones, la presencia de prurito asociado o el impacto psicológico del proceso, hace necesario el tratamiento.
La mayoría de las recomendaciones sobre el tratamiento están basadas en pequeñas series de casos, no existiendo a día de hoy suficiente evidencia para considerar ninguno de los tratamientos descritos un enfoque terapéutico universalmente aceptado.
Han sido publicadas pequeñas series de casos o algún caso aislado de diversos tratamientos, tanto tópicos como sistémicos, los cuales se enumeran y describen a continuación.
Tratamiento tópicoCorticoides tópicosLos esteroides tópicos son el tratamiento más comúnmente empleado en los casos publicados en la literatura. Reducen el prurito asociado y producen aclaramiento de las lesiones en algunas ocasiones2,4.
Los más empleados han sido los de media y alta potencia (clobetasol, metilprednisolona aceponato).
Inhibidores de la calcineurina tópicosSe han publicado casos de resolución de liquen aureus tras la aplicación de tacrolimus44 y pimecrólimus45 tópico durante varios meses.
Dada la cronicidad de las lesiones y la necesidad de regímenes terapéuticos prolongados, los inhibidores de la calcineurina los podemos considerar una buena alternativa a los corticoides tópicos, para conseguir aclaramiento o resolución de las lesiones de DPP.
FototerapiaLa fototerapia es una buena opción en el tratamiento de los casos extensos, o bien en aquellos que no han respondido a corticoides o inhibidores de la calcineurina tópicos.
Se ha postulado que la respuesta a la fototerapia sea debida a su efecto inmunomodulador, modificando la actividad de los linfocitos T y disminuyendo la producción de la IL 2, consiguiendo por este motivo la mejoría de las lesiones46.
El tratamiento con PUVA ha sido eficaz en pacientes con púrpura de Schamberg, dermatosis purpúrica liquenoide y liquen aureus. En las series publicadas hasta la fecha han sido necesarias entre 7 y 29 sesiones con dosis acumuladas de entre 16 y 49J/cm2, para conseguir la remisión de las lesiones. Los retratamientos también se han demostrado eficaces, y en algunos casos han sido necesarias dosis de mantenimiento durante meses para sostener la respuesta2,47–49.
La fototerapia UVB de banda estrecha (UVBbe) ha conseguido respuestas en las diversas formas clínicas de DPP, empleando entre 24 y 60 sesiones con dosis acumuladas de entre 11 y 49J/cm2. Al igual que con el tratamiento con PUVA en algunos casos han aparecido recidivas al suspender el tratamiento, pero igualmente con buena respuesta al retratamiento46,50,51.
El tratamiento con UVBbe debido a los escasos efectos secundarios que produce y la buena tolerancia del procedimiento, se considera una buena opción terapéutica, importante a tener en cuenta en los casos pediátricos, las formas extensas o que no hayan respondido a tratamientos tópicos5,52.
Tratamiento sistémicoPentoxifilinaSe han publicado algunos casos de respuesta de DPP a pentoxifilina oral. Se ha sugerido como mecanismo de acción, el efecto inhibidor de la pentoxiflina en la adherencia de los linfocitos T al endotelio vascular a través de la interacción con la molécula de adhesión celular (ICAM-1)53,54.
Se ha empleado en monoterapia, a dosis de 400mg dos o tres veces al día durante 2-3 meses42,43, o asociada a otros fármacos como prostaciclinas (PGI2)55 y corticoides orales56. Por otro lado, también existen publicaciones en las que se pone de manifiesto la ineficacia del tratamiento con pentoxiflilina en las DPP57.
Ácido ascórbico y bioflavonoides (rutina o rutósido)Basándose en el efecto que ejercen el ácido ascórbico y los bioflavonoides sobre el aumento en la síntesis de colágeno, reduciendo la permeabilidad vascular y por lo tanto mejorando las funciones de la barrera endotelial vascular, se han empleado altas dosis de vitamina C asociados a un glucósido flavonoide como es el rutósido o rutina, presente en los cítricos, durante varios meses, con mejoría del aspecto clínico de las lesiones y en algunos casos resolución de las mismas58.
OtrosExisten en la literatura publicaciones en las que presentan casos aislados con respuesta a diversos tratamientos sistémicos como griseofulvina59, colchicina60, metotrexato61 o ciclosporina62.
ConclusiónLas DPP son una entidad clínica frecuente en las consultas de dermatología y que muchas veces tienen una gran repercusión en la calidad de vida del paciente, tanto por la sintomatología como por sus repercusiones estéticas. Aunque son muy similares clínicamente existen una serie de características clínicas, histopatológicas y dermatoscópicas que permiten un diagnóstico más específico de la enfermedad.
Aunque la revisión de la literatura no aporte evidencia suficiente paran determinar un tratamiento de elección, sí contamos con distintas herramientas terapéuticas que pueden lograr diferencias significativas en estos pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Agradecemos al Dr. Valero Torres del Servicio de Anatomía patológica del Hospital Clínico Lozano Blesa la ayuda prestada en el análisis histopatológico.


