








DERMATOSIS EOSINOFILICAS DE LA EDAD PEDIATRICA
Eritema tóxico del recién nacido
El eritema tóxico neonatal es una dermatosis benigna y autolimitada que afecta aproximadamente a la mitad de los recién nacidos1. Suele aparecer entre las 24 y las 48 h de vida, pero ocasionalmente puede estar presente en el momento del nacimiento2. Se resuelve sin tratamiento en el curso de horas o días, aunque puede recidivar durante las primeras semanas de vida. Afecta por igual a ambos sexos y no tiene preferencia de razas.
Su causa es desconocida, aunque se supone debida a algún estímulo presente en el período neonatal que favorece la obstrucción del orificio pilosebáceo. Algunos autores proponen que se trate de una variedad peculiar de la enfermedad injerto contra huésped mediada por linfocitos maternos transferidos al niño durante el embarazo o el parto3.
El eritema tóxico neonatal puede afectar a toda la superficie cutánea, pero suele respetar plantas y palmas y ser más intenso en las zonas de oclusión. Se compone de máculas eritematosas, pápulas blancas o amarillentas, vesículas o pústulas sobre una base de eritema (fig. 1). No se acompaña de otros síntomas o signos, salvo eosinofilia periférica que aparece en el 15 % de los casos4. El estudio histológico muestra espongiosis eosinofílica, pústulas subcórneas e infiltrados perifoliculares de eosinófilos; las máculas pueden tener sólo un infiltrado escaso de eosinófilos en la dermis.
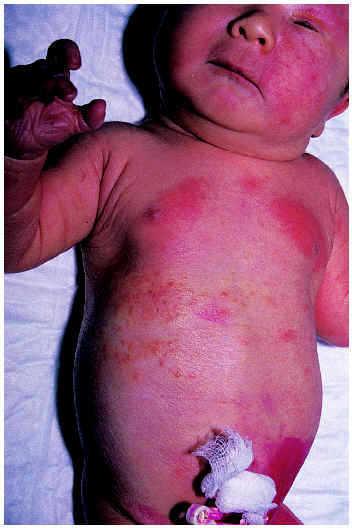
Fig. 1.--Eritema tóxico del recién nacido.
En las formas pustulosas, el estudio con hidróxido potásico (KOH) y el citodiagnóstico de Tzanck realizados con el contenido de una pústula serán muy útiles para descartar causas infecciosas. Asimismo, el predominio de eosinófilos facilita el diagnóstico de eritema tóxico neonatal frente a la melanosis pustulosa neonatal transitoria en la que predominan los neutrófilos. No obstante, esta última distinción tiene menor importancia, ya que la melanosis pustulosa neonatal transitoria es también banal y autorresolutiva, e incluso para algunos autores se trata de la misma entidad5.
Acropustulosis infantil
La acropustulosis infantil es una erupción vesiculopustulosa recidivante y pruriginosa que suele afectar a pies y manos durante la infancia. Generalmente aparece durante el primer año de vida, pero puede hacerlo más tarde. Sólo excepcionalmente está presente al nacer. Los brotes duran como máximo 15 días y se repiten cada 2 o 3 semanas. A medida que el niño crece los brotes duran menos y los intervalos libres de enfermedad se hacen más largos. La duración total de la enfermedad varía de meses a 2 o 3 años6,7.
Clínicamente se caracteriza por la aparición de vesículas y pústulas muy pruriginosas en manos y pies, sobre todo en las caras laterales (fig. 2). Excepcionalmente puede afectar también a partes no acras. El estudio citológico del contenido de las vesículas y pústulas muestra neutrófilos y a veces eosinófilos. La biopsia denota en fases iniciales necrosis de queratinocitos, posteriormente pústulas intraepidérmicas y, finalmente, pústulas subcórneas8. El contenido de estas pústulas es el descrito en la citología.
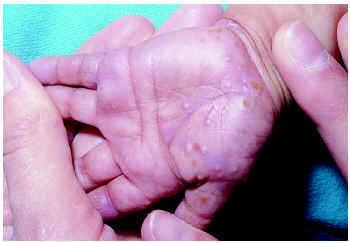
Fig. 2.--Acropustulosis infantil.
En muchos pacientes se recoge el antecedente personal o familiar de escabiosis, por lo que se ha propuesto que la acropustulosis infantil sea una reacción de hipersensibilidad peculiar a dicha infestación. No obstante, esta teoría no explicaría aquellos casos en los que no se recoge el antecedente de escabiosis9,10.
Como tratamiento se ha propuesto la dapsona, que reduce la duración de los brotes, así como la aplicación de corticoides tópicos que, por su efectividad y seguridad, pueden considerarse el tratamiento de elección7.
Pustulosis eosinofílica infantil
Lucky et al11 describieron en 1984 una peculiar pustulosis estéril pruriginosa y recidivante en el cuero cabelludo de lactantes. Aparece durante el primer año de vida, se han referido casos de inicio en el período neonatal12 y suele cursar con episodios recidivantes a lo largo de varios meses (fig. 3). En el estudio citológico del contenido de las pústulas se encuentran eosinófilos. Histopatológicamente existe un infiltrado inflamatorio mixto con numerosos eosinófilos alrededor y en los folículos pilosos. Los cultivos para bacterias y hongos son negativos. Puede cursar con leucocitosis y eosinofilia periférica13. Las lesiones suelen responder bien al tratamiento con esteroides tópicos14,15.
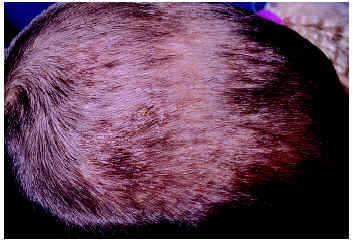
Fig. 3.--Pustulosis eosinofílica infantil del cuero cabelludo.
La pustulosis eosinofílica infantil se diferencia de la enfermedad de Ofuji por el grupo de edad afectado, su morfología de pústulas aisladas en vez de agrupadas en placas circinadas y su distribución preferente en cuero cabelludo y frente16. Existen autores que han planteado la posibilidad de que se trate de la misma enfermedad que la acropustulosis infantil17.
Eritema anular eosinofílico de la infancia
El eritema anular de la infancia fue descrito por Peterson y Jarratt18 en 1981 como una erupción generalizada urticariforme no pruriginosa, de causa desconocida, compuesta por lesiones arciformes o anulares recidivantes durante meses. Estos autores observaron que cada lesión individual persistía más de 24 h. La erupción era autorresolutiva sin secuelas en el curso de semanas o meses, en un niño sin otra enfermedad asociada. Histológicamente encontraron un infiltrado perivascular de linfocitos y eosinófilos. Peterson y Jarrat consideraron que la duración de cada lesión de casi 2 días, así como los infiltrados perivasculares más densos, eran suficientes para independizar esta entidad de la urticaria anular. Posteriormente, Toonstra y De Wit19 describieron un caso similar pero con lesiones sólo anulares, más grandes y más persistentes, pero también de resolución espontánea tras unos meses y con la misma histología descrita en el eritema anular de la infancia, y proponen un espectro clínico de dicha entidad. Los casos publicados posteriormente destacan la naturaleza asintomática, persistente y autorresolutiva del cuadro, así como la presencia de eosinófilos en el infiltrado20,21.
ENFERMEDADES EOSINOFILICAS DE HIPODERMIS, VASOS Y MUSCULOS
Paniculitis eosinofílica
La paniculitis eosinofílica, caracterizada por la infiltración septal o lobular del tejido adiposo por un gran número de eosinófilos (en la mayoría de casos más de 25 eosinófilos por campo de gran aumento) (fig. 4), se ha identificado en pacientes con una gran variedad de condiciones clínicas asociadas22,23. Así, para algunos autores, más que una entidad individualizada, la paniculitis eosinofílica sería un peculiar patrón histopatológico de paniculitis común a varias enfermedades24. El espectro clínico de lesiones cutáneas con cambios histológicos de paniculitis eosinofílica tiene como expresión más común las lesiones nodulares, aunque también incluye úlceras, pápulas y placas urticariales, púrpura, vesículas y pústulas, si bien todas ellas con un componente nodular subcutáneo añadido23. Las enfermedades asociadas con mayor frecuencia a paniculitis eosinofílica son la gnathostomiasis (lo que se ha conocido como paniculitis nodular migratoria eosinofílica), la vasculitis leucocitoclástica, el eritema nudoso y las picaduras de artrópodo. En ocasiones los eosinófilos infiltran también la dermis suprayacente con un aspecto histológico idéntico al de la celulitis eosinofílica de Wells e, incluso, la epidermis con aspecto de espongiosis eosinofílica23.
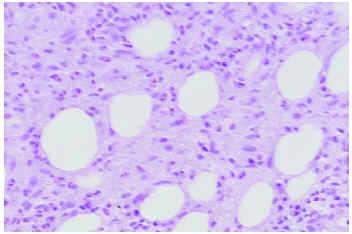
Fig. 4.--Paniculitis eosinofílica por picadura de artrópodo.
Se han descrito ejemplos de paniculitis eosinofílica como reacción local a una causa claramente identificable (picaduras de artrópodo, granulomas por inyección, medicamentos)23,25,26, si bien en la mayoría de los pacientes existe algún proceso cutáneo o sistémico asociado como dermatitis atópica y de contacto22,27, eritema nudoso22, celulitis eosinofílica de Wells22, enfermedades psiquiátricas (paniculitis facticia)22,28, diabetes29, enfermedades tiroideas22, infecciones (estreptococias, gnathostomiasis, toxocariasis, fascioliasis hepática)30-35, parotiditis recurrente36, vasculitis (vasculitis leucocitoclástica, poliarteritis nudosa)23, síndrome de Sjögren, paniculitis lúpica, morfea profunda, enfermedades hematológicas (leucemias, linfomas T y B, anemia refractaria con exceso de blastos) y neoplasias sólidas22,23.
Parece, pues, que la paniculitis eosinofílica es un hallazgo histológico poco específico que, no obstante, debe reconocerse con el fin de proceder a realizar los pertinentes exámenes clínicos y de laboratorio que permitan descartar las posibles enfermedades cutáneas y sistémicas asociadas24.
Vasculitis eosinofílica
Las enfermedades del tejido conjuntivo (artritis reumatoide, lupus eritematoso, síndrome de Sjögren) pueden cursar con un tipo de vasculitis diferente de las más habituales, las vasculitis neutrofílica o linfocítica37. De una serie de 98 pacientes con enfermedades del tejido conjuntivo y vasculitis necrosante cutánea, en ocho de ellos se trataba de una vasculitis eosinofílica38. Aunque aún menos frecuente, la vasculitis eosinofílica puede también ser idiopática39 o asociarse a síndrome hipereosinofílico40. Son pacientes que, de manera habitual, asocian artralgias, lesiones cutáneas, eosinofilia periférica e hipocomplementemia. La sintomatología cutánea se localiza con más frecuencia en las extremidades inferiores y consiste en pápulas eritematosas pruriginosas, placas urticariales, pápulas purpúricas y, ocasionalmente, microinfartos digitales y angioedema de cara y manos38-41.
Desde el punto de vista histopatológico, la vasculitis eosinofílica es una vasculitis de pequeño vaso, con necrosis fibrinoide de la pared, infiltrado por eosinófilos prominente y ausencia de leucocitoclasia. Los vasos afectados incluyen capilares, vénulas y arteriolas. Los estudios de inmunofluorescencia demuestran inmunoglobulina M (IgM) y C3 en las paredes vasculares y un abundante depósito angiocéntrico de la proteína básica mayor del eosinófilo, lo cual puede interpretarse como prueba de la implicación patogénica primaria del eosinófilo en el daño vascular38,42.
El curso suele ser benigno y, habitualmente, con buena respuesta a la corticoterapia sistémica. A diferencia del síndrome de Churg-Strauss no hay afectación de senos paranasales ni infiltrados pulmonares; además, en el caso del síndrome de Churg-Strauss, la vasculitis de vaso pequeño es neutrofílica y leucocitoclástica43,44.
Arteritis eosinofílica del cuero cabelludo
Grosshans y Asch45 han descrito el caso de una paciente fumadora que desarrolló episodios recurrentes de prurito, urticaria y eosinofilia periférica, junto a una peculiar tromboangitis eosinofílica de las arterias elásticas de mediano calibre del cuero cabelludo. A excepción de la eosinofilia persistente, el resto de los signos clínicos fueron autorresolutivos y no se comprobó ningún otro hallazgo clínico o biológico de enfermedad sistémica. Los autores proponen interpretar este cuadro como una entidad clinicopatológica individualizada, fácilmente distinguible de otras vasculopatías obliterantes (como la enfermedad de Buerger) o de vasculitis sistémicas (como el síndrome de Churg-Strauss) por la limitación y localización de los vasos afectados, así como por su naturaleza benigna y autorresolutiva sin necesidad de tratamiento.
Es posible que el cuadro descrito por Grosshans sea un trastorno bastante afín al que, previamente, se había denominado arteritis temporal juvenil. Ésta afecta a individuos jóvenes, en forma de lesiones nodulares o funiculares, indoloras, localizadas en cabeza y cuello, debidas a la oclusión de la luz arterial por proliferación intimal y densos infiltrados de linfocitos y eosinófilos intravasculares y extravasculares. Este tipo de tromboangitis obliterante se distingue fácilmente de la clásica arteritis de la temporal, desde el punto de vista clínico, por el grupo de edad afectado y la ausencia de complicaciones sistémicas del tipo de cefalea, mialgia, anemia, fiebre o pérdida de visión y, desde el punto de vista histopatológico, por la presencia de eosinófilos y ausencia de infiltrado granulomatoso y células gigantes46. Recientemente, se han discutido los posibles vínculos nosológicos entre la arteritis temporal juvenil y la enfermedad de Kimura47.
Miositis-perimiositis eosinofílica
En el concepto de miositis-perimiositis eosinofílica (MPE) se incluye un grupo de infrecuentes enfermedades musculares idiopáticas inflamatorias, asociadas a eosinofilia tisular y periférica, de curso clínico benigno, con buena respuesta terapéutica a antiinflamatorios o esteroides (AINE) y sin evidencia de afectación sistémica48. Los principales hallazgos histopatológicos son infiltrados inflamatorios en el perimisio con abundantes eosinófilos y, ocasionalmente, mionecrosis. El diagnóstico requiere la previa exclusión de parasitosis subyacente, síndrome de eosinofilia-mialgia, fascitis eosinofílica, vasculitis sistémicas con eosinofilia (síndrome de Churg-Strauss y panarteritis nudosa [PAN]), síndrome hipereosinofílico, reacciones farmacológicas y miopatías idiopáticas no eosinofílicas.
Recientemente, en una revisión de 26 casos publicados de MPE49, se ha señalado la presencia asociada de manifestaciones cutáneas en 10 de los casos (38 %). Éstas incluyen, por orden descendente de frecuencia, induración subcutánea profunda (edema sólido), eritema, urticaria-angioedema y lesiones papulosas eritematosas.
Es posible que algunos de los pacientes diagnosticados de síndrome hipereosinofílico, con lesiones cutáneas de angioedema y un pronóstico más favorable presenten, en realidad, diferentes enfermedades eosinofílicas benignas idiopáticas, como la MPE o el angioedema episódico con eosinofilia. Se trata de enfermedades limitadas a órganos específicos, como la piel o el músculo, y en las que no existen lesiones cardíacas asociadas mediadas por eosinófilos. No puede descartarse, sin embargo, que estas enfermedades representen, de hecho, el extremo benigno del espectro clínico del síndrome hipereosinofílico.
ENFERMEDADES EOSINOFILICAS ASOCIADAS CON FIBROSIS
Dentro de las enfermedades cutáneas esclerodermiformes, denominadas así porque se asocian a fibrosis dérmica y se asemejan a la esclerodemia, hay algunas que se acompañan de un infiltrado tisular de eosinófilos. Se incluyen dentro de este grupo tres dermatosis muy poco frecuentes y, para muchos autores, estrechamente relacionadas entre sí: la fascitis eosinofílica, el síndrome eosinofilia-mialgia y el síndrome del aceite tóxico. La activación de los eosinófilos, y una alteración secundaria en la regulación de la síntesis del colágeno en los fibroblastos, parece ser un mecanismo etiopatogénico común en estas entidades50,51.
Algunos autores han sugerido el término de «fascitis-paniculitis»52,53 para agrupar ciertas enfermedades que cursan con inflamación de la fascia y del tejido celular subcutáneo (septos fibrosos y lóbulos grasos). Este patrón histológico se encuentra en las tres entidades que nos ocupan y, además, en la morfea profunda y en la paniculitis lúpica52, en la enfermedad injerto contra huésped54, después del tratamiento radioterápico55, en determinadas infecciones56 y asociado a ciertas neoplasias57.
Fascitis eosinofílica de Schulman
En la fascitis eosinofílica o síndrome de Schulman, también denominada fascitis difusa con eosinofilia58, la afectación, aunque puede ser diseminada, está típicamente limitada a las extremidades y respeta, en general, las partes más distales. Es más frecuente en el sexo masculino y, en ocasiones, puede estar desencadenada de forma brusca por un esfuerzo físico inusual. Comienza con edema, eritema y dolor en la piel de las extremidades que evoluciona hacia la induración y la aparición de contracturas musculares que limitan la movilidad. La piel adquiere un característico aspecto abollonado (fig. 5) debido a la retracción del tejido celular subcutáneo entre los grupos musculares59,60. Suele acompañarse de eosinofilia en sangre periférica61, aumento de la velocidad de sedimentación globular (VSG) e hipergammaglobulinemia60.
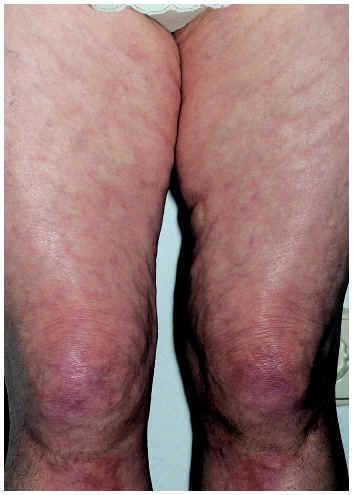
Fig. 5.--Fascitis eosinofílica. Aspecto cutáneo abollonado.
En el estudio histopatológico se observa un engrosamiento y una fibrosis de la fascia y de los septos del tejido celular subcutáneo, acompañados de un infiltrado polimorfo, con linfocitos, células plasmáticas, mastocitos y eosinófilos. En ocasiones, los eosinófilos son muy numerosos, pero lo más habitual es que sólo aparezcan aumentados focalmente62. En las fases avanzadas, la afectación se extiende a la dermis profunda y los hallazgos histológicos son indistinguibles de los de la esclerodermia.
En relación con lo anterior, algunos autores, consideran a la fascitis eosinofílica como una forma de esclerodermia localizada y profunda63-65. Sin embargo, y a diferencia de ésta, el fenómeno de Raynaud y la afectación de órganos internos son muy raros66, aunque se ha descrito algún caso aislado de afectación renal67. Además, suele responder al tratamiento precoz con corticoides orales e, incluso ocasionalmente, la remisión puede ser espontánea68,69. Se ha descrito su asociación con síndromes mieloproliferativos, linfomas de células T70,71 y con distintas enfermedades del tejido conjuntivo72,73, y se ha implicado a la espiroqueta Borrelia burgdorferi en su etiopatogenia74.
Síndrome eosinofilia-mialgia
El síndrome eosinofilia-mialgia es otro cuadro clínico esclerodermiforme que se observó por primera vez en 1989 como consecuencia de la ingesta de complejos vitamínicos y fármacos que contenían L-triptófano75,76. Los pacientes afectados por este síndrome presentan lesiones cutáneas similares a las descritas en la fascitis eosinofílica junto con mialgias y habitualmente una marcada eosinofilia periférica77,78. A diferencia de la fascitis eosinofílica, este síndrome cursa con frecuencia, con un exantema pruriginoso, fiebre, polineuropatía y afectación visceral (encefalopatía, neumonitis, miocarditis, etc.) que incluso puede ser mortal79,80. En el estudio histopatológico se observa un infiltrado inflamatorio linfohistiocitario que puede afectar además de a la fascia y al tejido celular subcutáneo, a todo el espesor de la dermis81. La eosinofilia tisular suele ser poco llamativa82.
Síndrome del aceite tóxico
El denominado síndrome del aceite tóxico surgió en 1981 en algunas regiones de España, a partir de una intoxicación epidémica por aceite de colza adulterado83. Este síndrome tiene dos fases distintas en su curso evolutivo. La fase aguda se caracteriza por la aparición de un exantema escarlatiniforme o morbiliforme, pruriginoso, acompañado de neumonitis, síntomas gastrointestinales, mialgias y eosinofilia. Varios estudios han puesto de manifiesto el posible papel patogénico del eosinófilo, sobre todo en la fase aguda de la enfermedad, al demostrar la infiltración de distintos tejidos por estas células84. La fase crónica aparece a partir del quinto o sexto mes del comienzo de la enfermedad. Durante ella se observan alteraciones neuromusculares (atrofia muscular, pérdida de la sensibilidad), acompañadas en algunos pacientes por unas lesiones cutáneas que pueden adquirir una gran similitud clínica e histopatológica con la fascitis eosinofílica y la esclerodermia85. Aparecen en la piel zonas de edema e induración que pueden ser localizados o afectar a gran parte del tegumento y que pueden llevar a la aparición de importantes contracturas articulares y deformidades. En el estudio histopatológico se observa una marcada fibrosis que alcanza el tejido celular subcutáneo junto con atrofia de las glándulas sudoríparas y del epitelio folicular. También en esta fase la afectación visceral (esofágica, pulmonar y ocasionalmente renal) puede ser importante y condicionar el pronóstico de esta enfermedad86.
Más de 20 años después de su descripción inicial, siguen siendo numerosos los estudios que pretenden identificar el agente causal y esclarecer la patogenia de esta enfermedad, con el objetivo de poder prevenir nuevas enfermedades similares que pudieran surgir en el futuro87-89.
Otras dermatosis con eosinofilia tisular (eosinofilia secundaria)
Existen otras muchas dermatosis que también se acompañan de un infiltrado tisular de eosinófilos, pero que, debido a su gran heterogeneidad, resultan difíciles de clasificar en un grupo común (tabla 1). Se han denominado dermatosis con eosinofilia tisular secundaria, ya que en ellas la presencia de eosinófilos suele ser un hallazgo secundario, si bien en ocasiones constituye un dato clave para poder orientar el diagnóstico (enfermedades parasitarias, reacciones a drogas). Aunque de forma inconstante, estas enfermedades se acompañan también de eosinofilia periférica. Además, es bien conocido el papel patógeno del eosinófilo y de sus proteínas citoplasmáticas en alguna de las dermatosis de este grupo, como toxicodermias90, dermatitis atópica90-92, urticaria93,94, penfigoide ampolloso90,95, incontinentia pigmenti96.

Los eosinófilos pueden hallarse bien formando parte más o menos importante del infiltrado inflamatorio de la dermis, bien invadiendo la epidermis. Cuando esta exocitosis de eosinófilos se acompaña de edema intercelular (espongiosis), estamos ante el patrón histológico denominado espongiosis eosinofílica, término propuesto por Emerson y Wilson-Jones97 en 1968, en su descripción inicial a propósito de 7 pacientes con pénfigo. La espongiosis eosinofílica es un hallazgo prácticamente constante en ciertas enfermedades como la incontinentia pigmenti (fase vesiculoampollosa), el eritema tóxico neonatal o la foliculitis pustulosa eosinofílica (afectando la pared del infundíbulo folicular). Además, puede encontrarse con una frecuencia variable en gran cantidad de dermatosis98-101; entre ellas destacan las enfermedades ampollosas autoinmunes, fundamentalmente las distintas formas de pénfigo97,102-105 y el penfigoide ampolloso106, en las cuales la espongiosis eosinofílica suele observarse en sus fases iniciales y, en ocasiones, como único hallazgo histopatológico100 (tabla 2).

Dentro de este grupo de dermatosis con eosinofilia tisular secundaria, hay algunas enfermedades de reciente descripción que presentan ciertas características peculiares y que se comentan brevemente a continuación.
Dermatosis eosinofílica de las enfermedades mieloproliferativas
Se trata de una término acuñado por Byrd et al107 en 2001 en referencia a una erupción cutánea papulosa o vesiculoampollosa, pruriginosa y resistente a los tratamientos habituales (fig. 6), que aparece en pacientes con discrasias o neoplasias hematológicas y que se caracteriza histopatológicamente por un infiltrado linfohistiocitario con abundantes eosinófilos en dermis superficial y profunda. Para poder establecer este diagnóstico deben excluirse otras causas conocidas de eosinofilia tisular. Su etiopatogenia es desconocida y la fototerapia con luz ultravioleta B de banda estrecha debe considerarse la primera alternativa terapéutica, si bien muchas veces los resultados son desalentadores.
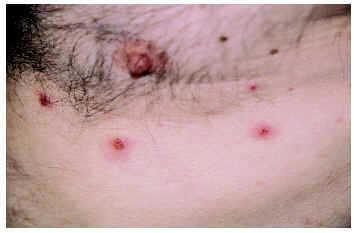
Fig. 6.--Dermatosis eosinofílica de las enfermedades mieloproliferativas.
Esta entidad se corresponde probablemente con la, hasta entonces, denominada reacción exagerada a picaduras de insectos o artrópodos, descrita también en pacientes con neoplasias hematológicas y que recibe este nombre debido a su similitud clínica e histopatológica (abundancia de eosinófilos) con las picaduras de artrópodos108,109. Sin embargo, y debido a que en la mayor parte de los casos no puede recogerse el antecedente de picaduras en la anamnesis, esta denominación parece menos apropiada. Además, y teniendo en cuenta que esta dermatosis, incluso en el trabajo de Byrd, aparece no sólo en las enfermedades mieloproliferativas, sino también en otros tipos de neoplasias hematológicas como la leucemia linfocítica crónica107,108,110, creemos que quizá sería aún más correcto hablar de dermatosis eosinofílicas de las enfermedades o neoplasias hematológicas.
Erupción eosinofílica polimorfa y pruriginosa asociada a radioterapia
Es una dermatosis descrita en 1999 por Rueda et al111 y que, en su serie, aparece en el 17 % de 83 pacientes con cáncer durante el tratamiento con radioterapia. Consiste en la aparición de una erupción cutánea polimorfa (pápulas eritematosas, vesículas o ampollas y excoriaciones), localizada fundamentalmente en las extremidades inferiores, y acompañada de un prurito que suele ser generalizado. En ocasiones estas lesiones son persistentes, incluso una vez terminado el tratamiento. En un caso, reflejado recientemente en la literatura112, la erupción surgió varios meses después de finalizada la radioterapia. El estudio histopatológico muestra la presencia de un infiltrado con numerosos eosinófilos en la epidermis, dermis (intersticial y perivascular) y tejido celular subcutáneo. A diferencia del penfigoide ampolloso, con el que histológicamente hay que establecer el diagnóstico diferencial, los estudios de inmunofluorescencia revelan depósitos de IgM y C3 perivasculares y no en la unión dermoepidérmica.
Foliculitis eosinofílica asociada a la infección por virus de la inmunodeficiencia humana
Aunque la asociación entre la foliculitis pustulosa eosinofílica y la infección por el virus de la inmunodeficiencia humana (VIH) ya había sido descrita con anterioridad113, a partir del trabajo de Rosenthal et al114 en 1991 se tiende a considerar a la foliculitis eosinofílica asociada a la infección por VIH como una entidad clínica diferenciada de la enfermedad de Ofuji y que comparte únicamente con ésta sus características histopatológicas. Se caracteriza clínicamente por la presencia de múltiples pápulas foliculares de curso crónico y localizadas en la cabeza, tronco y raíz de miembros. Además, y también a diferencia de la enfermedad de Ofuji, el prurito está siempre presente y no existe leucocitosis; al contrario, el recuento de leucocitos puede estar disminuido y el número de linfocitos CD4 suele ser menor de 300/μl, reflejo en estos pacientes de un estado de inmunodepresión. La fototerapia con luz ultravioleta B es efectiva para su tratamiento, pero se requiere un tiempo prolongado, por lo que debe ser manejada con precaución en este tipo de pacientes inmunodeprimidos. Una alternativa terapéutica la constituyen los corticoides tópicos de alta potencia.
Pustulosis exantemática aguda generalizada, variedad eosinofílica
Descrita por Deylot et al115 en 1980, consiste en la aparición brusca de un exantema pustuloso diseminado, acompañado de fiebre y neutrofilia, con tendencia a la autoinvolución y que tiene como causa más frecuente los fármacos, principalmente los antibióticos. Desde el punto de vista histopatológico se caracteriza por la presencia de pústulas neutrofílicas espongiformes superficiales, acompañadas de un importante edema de la dermis superior y, ocasionalmente, necrosis de queratinocitos, infiltrado perivascular de eosinófilos y vasculitis116. Sin embargo, recientemente, Herrera et al117 han descrito un caso el que las pústulas estaban constituidas fundamentalmente por eosinófilos, en lugar del habitual neutrófilo, lo cual se interpretó como una variante eosinofílica de esta entidad.
ENFERMEDADES ASOCIADAS A EOSINOFILIA PERIFÉRICA
Angioedema episódico con eosinofilia o síndrome de Gleich
El angioedema episódico con eosinofilia fue descrito por primera vez en 1984 por Gleich et al118 y se caracteriza por la asociación de angioedema recurrente, urticaria, fiebre y concentraciones séricas elevadas de IgM. Durante los ataques de esta enfermedad se produce un aumento del peso corporal y una leucocitosis importante (de hasta 100.000/μl) correspondiendo incluso más del 50 % de estos leucocitos a eosinófilos119-122. En caso de realizar estudio histológico de una zona de angioedema se encuentra edema dérmico y un infiltrado difuso perivascular de linfocitos, histiocitos y eosinófilos, generalmente desgranulados. Los corticoides por vía sistémica logran una rápida remisión de los síntomas pero no previenen las recidivas, por lo cual resulta muchas veces necesario realizar un tratamiento de mantenimiento con dosis bajas119. El síndrome de Gleich, si bien tiene características comunes con el síndrome hipereosinofílico, se diferencia de éste por su carácter cíclico y su buen pronóstico debido a la ausencia de afectación visceral.
En los últimos años autores japoneses123-125 han descrito, en mujeres orientales jóvenes, un tipo de angioedema de carácter no episódico y con diferencias clínicas y evolutivas con respecto al inicialmente descrito por Gleich. Por ello, han propuesto la clasificación del angioedema con eosinofilia en dos tipos: angioedema recurrente (tipo Gleich) y angioedema no recurrente.
La etiopatogenia del síndrome de Gleich es desconocida. Probablemente la eosinofilia periférica y de los tejidos119,126,127 sea secundaria a la activación de linfocitos T por estímulos desconocidos. La liberación de distintas interleucinas (IL-2, IL-5) sería responsable del reclutamiento y la activación de los eosinófilos que, tras la desgranulación y liberación de su contenido citotóxico, provocarían las lesiones tisulares. Kawano et al128 han señalado que los eosinófilos del angioedema cíclico con eosinofilia no expresan el marcador CD-69 en la superficie celular (un marcador de actividad celular) y han propuesto que su ausencia podría explicar el carácter benigno de esta enfermedad, a diferencia de lo que sucede en el síndrome hipereosinofílico.
Síndrome hipereosinofílico
El síndrome hipereosinofílico es una enfermedad multisistémica caracterizada por eosinofilia en sangre periférica e infiltración de eosinófilos en distintos órganos129,130. Inicialmente descrito por Hardy y Anderson131 en 1968, Chusid et al132 establecieron posteriormente los siguientes criterios diagnósticos: a) eosinofilia en sangre periférica superior a 1.500/μl de al menos 6 meses de duración; b) ausencia de cualquier causa reconocida de eosinofilia, y c) lesiones viscerales secundarias a la infiltración de los tejidos por eosinófilos.
El síndrome hipereosinofílico afecta a varones con más frecuencia que a mujeres, en una proporción 9:1133,134. Es más común en la edad adulta, aunque se ha descrito en todos los grupos de edades, incluyendo la infancia y la adolescencia133,135. Las manifestaciones clínicas más frecuentes están relacionadas con la afectación cardíaca, hematológica, neurológica, gastrointestinal, pulmonar, renal y cutánea.
La afectación cutánea es frecuente (más del 50 % de los casos) y polimorfa. Ninguna expresión clínica es específica. Las lesiones más frecuentes son pápulas y nódulos eritematosos y pruriginosos, o angioedema y urticaria con dermografismo136. Los pacientes con angioedema y urticaria suelen presentar un curso benigno de la enfermedad, sin complicaciones cardíacas y neurológicas y con buena respuesta al tratamiento136. Por el contrario, la aparición de ulceraciones mucosas bucogenitales, así como oculares y digestivas, parece estar relacionada con formas de mal pronóstico137. Otras manifestaciones cutáneas menos frecuentes relacionadas con el síndrome hipereosinofílico incluyen: microtrombos vasculares, vasculitis138, necrosis digital, lesiones vesiculoampollosas, eritrodermia, eritema anular centrífugo139, eritema elevatum diutinum, prurito acuagénico, alopecia y distrofia ungueal. Se ha descrito algún ejemplo de síndrome hipereosinofílico precediendo al desarrollo de papulosis linfomatoide y linfoma de células T140. En ocasiones, la afectación cutánea por el síndrome hipereosinofílico puede ser aislada sin que, tras un estudio completo de extensión, sea posible encontrar ninguna otra anomalía visceral141,142.
La histología de las lesiones cutáneas resulta poco específica. En general, consiste en un infiltrado inflamatorio mixto perivascular de células mononucleares y eosinófilos136.
La etiopatogenia sigue siendo desconocida, pero cada vez más argumentos implican a los linfocitos T en la responsabilidad de la eosinofilia. Se han demostrado niveles séricos elevados del receptor soluble de la IL-2143 y, sobre todo, una proliferación clonal de linfocitos T CD3 y CD4+ de fenotipo Th2, secretora de IL-2, IL-4, IL-5 e IL-13144.
El objetivo del tratamiento en el síndrome hipereosinofílico es mantener la cifra de eosinófilos por debajo de 1.000/μl. Se ha propuesto una clasificación terapéutica en 4 grupos, según el grado de afectación clínica y biológica, para cada uno de los cuales la actitud varía desde la abstinencia terapéutica (sólo seguimiento clínico y analítico cada 3-6 meses) en pacientes con afectación limitada a un solo órgano (con excepción del corazón y el sistema neurológico), hasta la necesidad de tratamientos agresivos con corticoides asociados a citostáticos en pacientes con afectación multisistémica y alteraciones hematológicas con progresión leucémica145.
COMENTARIO
Las enfermedades cutáneas caracterizadas por infiltrados eosinofílicos representan un grupo patológico heterogéneo en el que no parece fácil encontrar factores comunes que promuevan una interpretación global146,147. Desde el punto de vista clínico, comprenden tanto dermatosis generalizadas como lesiones localizadas, y su expresividad varía desde un simple edema, formación de vesículas y ampollas, lesiones eccematosas, erupciones papulosas o cambios fibromatosos de tejidos profundos. En muchos de los casos, existe prurito más o menos intenso como síntoma acompañante148. La heterogeneidad caracteriza, también, los hallazgos histopatológicos; así, el infiltrado eosinofílico puede involucrar preferentemente la epidermis (enfermedad de Ofuji), la dermis (celulitis de Wells, granuloma facial), o tejidos blandos más profundos como hipodermis, fascia o músculo (morfea, fascitis eosinofílica, úlcera eosinofílica)148. Además, la eosinofilia puede ser sólo tisular local (picaduras de insecto), sólo sanguínea sin infiltración tisular (algunas reacciones medicamentosas), o en ambas localizaciones (penfigoide, celulitis de Wells)149.
Uno de los argumentos esgrimidos a favor de la visión integradora aceptada para las dermatosis neutrofílicas150 consiste en la existencia de cuadros mixtos o de superposición en los cuales resulta difícil discriminar entre una u otra de las posibles enfermedades del grupo, tal como ocurre, por ejemplo, entre el pioderma gangrenoso superficial ampolloso y formas atípicas del síndrome de Sweet. Tal circunstancia no ocurre entre las enfermedades del grupo de las dermatosis eosinofílicas; cada una de ellas posee unas características clinicopatológicas distintivas, no existen ejemplos de cuadros de superposición y no parece justificable su interpretación como el espectro de una pretendida única enfermedad.
Por lo tanto, para el conjunto de dermatosis eosinofílicas sólo resulta común la naturaleza de la célula efectora inmunitaria predominante y, en relación directa con ello, la eventual asociación coincidente con ciertos factores etiopatogénicos conocidos determinantes de eosinofilia. Así pues, no puede resultar extraño que unos mismos agentes, inductores de una respuesta inmunitaria con hipereosinofilia, puedan aparecer en relación con varios de los tipos de enfermedad cutánea por eosinófilos. Se ha descrito, incluso, algún ejemplo de coexistencia de varias enfermedades cutáneas eosinofílicas, como la foliculitis pustulosa eosinofílica y la enfermedad de Kimura o la celulitis eosinofílica151.
Resulta interesante, también, recordar que los infiltrados eosinofílicos cutáneos son habitualmente infiltrados mixtos en los que, junto a los eosinófilos, están presentes también linfocitos, monocitos o neutrófilos148. Este hecho contrasta con los infiltrados eosinofílicos esencialmente monomorfos que ocurren en otros órganos, como, por ejemplo, los encontrados en una mucosa intestinal con oxiuriasis. Parece por lo tanto que, en la piel especialmente, los múltiples sistemas efectores de la inmunidad actúan como una red intercoordinada. La presencia de eosinófilos puede interpretarse, así, sólo como una más de las consecuencias del patrón de respuesta inmunitaria con predominio del linfocito Th2, en contraste con el patrón Th1 en que son atraídos neutrófilos y macrófagos como células efectoras152.
Las dermatosis eosinofílicas, por lo tanto, reflejan fenómenos de reactividad inmunitaria, local o sistémica, frente a conocidos antígenos externos (helmintos, proteínas de artrópodos, medicamentos) o autoantígenos (penfigoide, neoplasias), si bien en la mayoría de las enfermedades clásicas el agente causal sigue siendo desconocido153. Para comprender adecuadamente la etiopatogenia de cada una de estas enfermedades será obligado tanto conocer los antígenos implicados, como entender mejor la función efectora del eosinófilo en las reacciones inflamatorias cutáneas, su papel modulador y las circunstancias en que, por el contrario, predominan sus efectos citotóxicos generadores de enfermedad149.


