











INTRODUCCIÓN
Conocer significa, en buena medida, clasificar. Así, la especialidad de Dermatología no se desarrolló como una auténtica ciencia hasta que a finales del siglo XVIII se elaboró el alfabeto morfológico de las lesiones elementales como herramienta para definir con exactitud y clasificar las enfermedades observadas. El incremento de conocimientos que caracteriza el progreso médico se suele reflejar en el incremento de la complejidad y epígrafes dentro de cualquier clasificación. Sin embargo, en una actitud que los filósofos considerarían dialéctica, resulta en ocasiones conceptualmente útil intentar realizar labores de síntesis más que de desglose. Un ejemplo afortunado de visión patogénica integradora lo constituye el concepto de dermatosis neutrofílicas1, 2. Por analogía, lo que se pretende en este trabajo es realizar una revisión conjunta de las dermatosis eosinofílicas (DE), entendiendo por tales todas aquellas enfermedades cutáneas en cuyo examen dermatopatológico los eosinófilos constituyen un hallazgo diagnóstico esencial. Se revisarán especialmente las enfermedades cutáneas primarias con eosinofilia tisular idiopática, que puede o no ir acompañada de una eosinofilia periférica. Sólo se hará mención más breve de las enfermedades cutáneas de etiología conocida (enfermedades parasitarias, reacciones a fármacos, etc.) en las que la eosinofilia tisular es un hallazgo secundario y de aquellas otras definidas más bien por la eosinofilia en sangre periférica. Sobre la posible unidad patogénica de este grupo de enfermedades, en analogía a la visión integradora aceptada para las dermatosis neutrofílicas, se discutirá en la parte final de la revisión.
EL EOSINOFILO
Paul Ehrlich en 1879 denominó eosinófilo a esta célula debido a la avidez con la que se tiñen sus gránulos citoplásmicos con el colorante ácido eosina. El eosinófilo se desarrolla a partir de las células pluripotenciales de la médula ósea, permanece unas 24 horas en la circulación sanguínea y migra principalmente hacia pulmón, intestino y tracto genitourinario, con una supervivencia en los tejidos de varios días3. Los individuos sanos poseen pocos eosinófilos circulantes (menos del 2%-10% de los leucocitos circulantes), ya que no es una célula esencialmente sanguínea, sino que alcanza sus mayores concentraciones en los tejidos. Es importante conocer estos flujos ya que, en el caso de patologías crónicas, es posible encontrar eosinofilia tisular importante y eosinofilia sanguínea leve o inexistente.
Los gránulos citoplásmicos del eosinófilo, específicos y distintivos, contienen 4 proteínas catiónicas que son citotoxinas responsables de la mayor parte de sus funciones: la proteína básica mayor (MBP), la peroxidasa de los eosinófilos (EPO), la neurotoxina derivada de los eosinófilos (EDN) y la proteína catiónica de los eosinófilos (ECP). Estos gránulos muestran al microscopio electrónico un centro electrón-denso y una matriz menos densa. La MBP constituye el centro de los gránulos, mientras que la EPO, EDN y ECP constituyen la matriz de los mismos. La MBP, además de ser una potente toxina, induce la liberación de histamina a partir de los basófilos y mastocitos y activa los neutrófilos y las plaquetas3.
En cuanto a sus funciones, el eosinófilo es una célula efectora de la respuesta inmunitaria en la defensa antiparasitaria y con actividad antitumoral. Tiene actividad fagocítica, puede ingerir microorganismos y destruirlos, pero su papel esencial es la eliminación de blancos biológicos como los helmintos, protozoos, bacterias y células tumorales. El eosinófilo participa en la defensa inmune de las mucosas y, además, se ha comprobado in vitro que posee potente actividad antivírica, con inhibición de infecciones por virus ARN4. En relación con ello, son numerosas las situaciones que pueden dar lugar a un aumento de eosinófilos en los tejidos y/o sangre periférica, bien formando parte del patrón diagnóstico histológico, bien participando en la patogenia de la enfermedad únicamente a través del depósito de las proteínas de sus gránulos.
CLASIFICACION DE LAS DERMATOSIS EOSINOFILICAS
En la bibliografía dermatológica apenas existen revisiones de conjunto a propósito de las DE5, 6 y, por tanto, son escasos los intentos de clasificación de las mismas. Se encuentran eosinófilos cutáneos en circunstancias tan diversas que resulta difícil establecer una clasificación exhaustiva y lógica de todas las posibles dermatosis implicadas.
En la tabla 1 se recoge la clasificación que proponemos. Se consideran 5 DE primarias mayores: la foliculitis pustulosa eosinofílica (FPE), la celulitis eosinofílica (CE), la hiperplasia angiolinfoide con eosinofilia (HALE), el granuloma facial (GF) y la úlcera eosinofílica de la mucosa oral (UE) interpretada en el sentido de expresión mucosa de infiltrados eosinofílicos autoinvolutivos de posible topografía diversa (dermatosis eosinofílicas mucocutáneas autoinvolutivas). Se hace mención separada a dos cuadros infrecuentes y de discutible ubicación nosológica: la papuloeritrodermia y la dermatosis eosinofílica paquidérmica. El resto de DE se agrupan en los siguientes epígrafes: DE de la edad pediátrica, infiltrados eosinofílicos de hipodermis, vasos y músculos, enfermedades eosinofílicas asociadas con fibrosis, otras dermatosis con eosinofilia tisular (eosinofilia tisular secundaria) y enfermedades asociadas a eosinofilia periférica.

FOLICULITIS PUSTULOSA EOSINOFILICA
Isé y Ofuji7 describieron en 1965 una dermatosis que, inicialmente, interpretaron como una variante folicular de la dermatosis pustulosa subcórnea, si bien la reinterpretaron poco después como una dermatosis individualizada8. Desde 1974 se viene empleando la denominación de enfermedad de Ofuji9.
Características clínicas
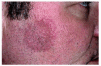
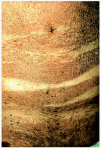
Las manifestaciones clínicas de la FPE consisten en la aparición de brotes recurrentes de papulopústulas foliculares estériles, agrupadas en áreas bien delimitadas, configurando placas de morfología circinada (figs. 1 y 2) que tienden a la progresión centrífuga y a la involución central10, 11. La distribución preferente de las lesiones es en las zonas más seborreicas de la piel: cara, espalda y zona más proximal de la superficie de extensión de las extremidades superiores12-18. La enfermedad predomina en hombres, en la tercera o cuarta décadas de la vida, y se ha descrito con más frecuencia en la raza oriental. El prurito es un síntoma frecuente pero no constante. Se suele acompañar de leucocitosis y eosinofilia periférica mayor del 5%. Los cultivos bacterianos y micológicos siempre resultan negativos19, 20.
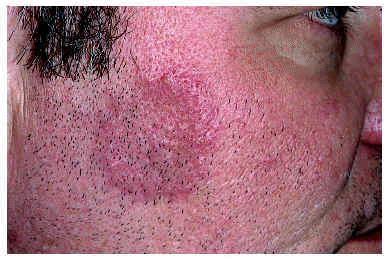
Fig. 1.--Foliculitis pustulosa eosinofílica: placa circinada en una mejilla.
Fig. 2.--Foliculitis pustulosa eosinofílica: pústulas foliculares en la periferia de una lesión.
Hallazgos histopatológicos
Desde el punto de vista histopatológico, en las etapas iniciales se observa espongiosis en la pared del infundíbulo folicular a expensas de un infiltrado de eosinófilos y células mononucleares. En etapas avanzadas del proceso se puede apreciar un auténtico clivaje o vesícula longitudinal, con neutrófilos, linfocitos y gran número de eosinófilos, que separa toda la pared infundibular (fig. 3) y afecta, con frecuencia, a la glándula sebácea (fig. 4)21. Algunas pústulas, primariamente intrafoliculares, se extienden secundariamente desde el infundíbulo hacia la epidermis vecina formando pústulas epidérmicas subcórneas10. Es posible encontrar, además, algún grado de degeneración mucinosa de la vaina epitelial externa22, 23.
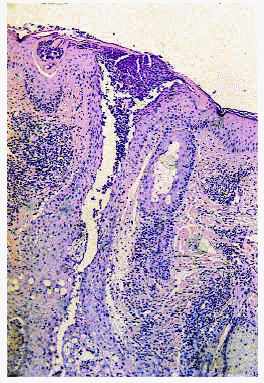
Fig. 3.--Foliculitis pustulosa eosinofílica: absceso infundibular.
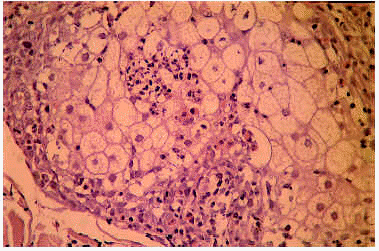
Fig. 4.--Foliculitis pustulosa eosinofílica: infiltración eosinofílica de la glándula sebácea.
Etiopatogenia
La etiología de la FPE es desconocida. La enfermedad se puede interpretar como una reacción de hipersensibilidad centrada en el folículo a estímulos antigénicos de localización folicular en un terreno de disregulación inmune que favorece el protagonismo del eosinófilo como célula efectora final24, 25. Entre las hipótesis fisiopatológicas para explicar esta enfermedad, se ha especulado sobre un cierto papel de la actividad sebácea debido a la distribución de las lesiones, los habituales antecedentes personales de acné y los cambios histopatológicos que respetan la porción inferior del folículo por debajo de la glándula sebácea11. De hecho, en lípidos de la superficie cutánea obtenidos de áreas seborreicas de los enfermos se ha demostrado la existencia de factores quimiotácticos para los eosinófilos26.
Se han comunicado ejemplos, interpretados como FPE, en los cuales se pudo establecer relación con agentes etiológicos específicos. Se trataría, en estos casos, de un patrón de respuesta inflamatoria exagerada a saprófitos o patógenos foliculares como Demodex folliculorum, dermatofitos27, 28, Pseudomonas aeruginosa29 o larva migrans30. También se ha sugerido la posible implicación en la enfermedad de un retrovirus no identificado31. La foliculitis eosinofílica asociada a la infección por virus de la inmunodeficiencia humana (VIH)32, 33 y la foliculitis eosinofílica infantil34 hoy se tienden a considerar entidades clínicas individualizadas que únicamente comparten con la enfermedad de Ofuji sus características histopatológicas.
No es infrecuente encontrar la FPE en un terreno de disregulación inmune. Se han descrito anomalías en las inmunoglobulinas, defectos en la movilidad de los neutrófilos y alteraciones en las funciones supresoras de las células T35-37. Entre las posibles patologías asociadas no resulta rara la asociación con malignidad hematológica, como síndrome mielodisplásico38, linfoma no Hodgkin36, 39, leucemia linfocítica crónica40, leucemia mieloide aguda, mieloma y macroglobulinemia de Waldenström41. Se han comprobado elevaciones en los niveles de citocinas de los linfocitos Th2 (IL-5, quimiotáctica para eosinófilos) en los pacientes con FPE y, por el contrario, las citocinas de origen Th1 (interferón gamma) están descendidas42, 43. En algunos trabajos se ha demostrado la existencia de inmunoglobulinas circulantes dirigidas contra el citoplasma de las células basales de la vaina epitelial externa folicular44 o contra la sustancia intercelular45, lo cual vincularía la enfermedad con fenómenos de autoinmunidad.
Pronóstico y tratamiento
La evolución habitual de la FPE es crónica, con exacerbaciones y remisiones a lo largo de un período variable de tiempo que llega, en casos de evolución prolongada, a varios años. Los tratamientos que, hasta el momento, se han comunicado útiles en la FPE incluyen corticosteroides orales y tópicos, cetirizina (con acción antieosinofílica)46, colchicina, dapsona47, 48, minociclina, cetotifeno, oxifenbutazona, indometacina (un inhibidor de síntesis de metabolitos del ácido araquidónico, como el LT-B4, quimiotácticos para eosinófilos)49, 50, isotretinoína (para la inhibición de factores quimiotácticos cuya presencia se sospecha en los lípidos sebáceos)51, acitretino (como inductor de maduración de los queratinocitos foliculares activados en la FPE)52, 53, fototerapia UVB54, psoraleno y luz ultravioleta (PUVA), ciclosporina55, interferón gamma e interferón-alfa 2b (por el efecto inhibitorio de la función de los Th2 que segregan IL-5)56-58. Destacaremos, por su especial predicamento, los esteroides sistémicos, la sulfona y la indometacina59. Con todo, algunos pacientes parecen no responder a ningún régimen terapéutico. Existen algunos casos comunicados de evolución fatal per se 60 o por la patología asociada39.
CELULITIS EOSINOFILICA
El síndrome de Wells o celulitis eosinofílica fue descrito por Wells en 1971 como dermatitis granulomatosa recurrente con eosinofilia61. Se trata de una rara dermatosis inflamatoria, de carácter recidivante y etiología desconocida62.
Manifestaciones clínicas
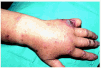
La CE se caracteriza clínicamente por la aparición de lesiones que simulan una celulitis bacteriana, de modo recurrente y con una evolución en general benigna. En el curso clínico del síndrome de Wells se describen dos fases. En la inicial, o celulítica, aparecen de forma súbita múltiples placas edematosas y eritematosas, bien definidas, que se extienden en el curso de días. Ocasionalmente pueden aparecer vesículas y ampollas en esta fase, del mismo modo que aparecen en las auténticas erisipelas (figs. 5 y 6). En la segunda fase, o granulomatosa, se produce un aclaramiento central de las lesiones, con persistencia de un borde rosado o violáceo durante semanas63. Las lesiones al curar pueden dejar hiperpigmentación, o incluso atrofia. Generalmente no hay afectación sistémica, aunque se han descrito casos con fiebre o artralgias. Con frecuencia aparece eosinofilia en sangre periférica durante el brote, pero su presencia no es imprescindible para el diagnóstico. Estos episodios pueden repetirse durante años, con períodos de remisión y de exacerbación de duración variable. Este tipo de celulitis puede aparecer a cualquier edad, incluso de forma congénita62.
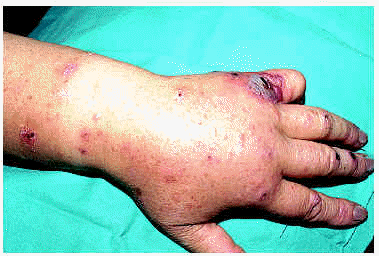
Fig. 5.--Celulitis eosinofílica en dorso de mano.
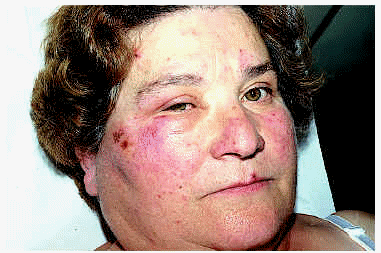
Fig. 6.--Celulitis eosinofílica en una mejilla.
El diagnóstico diferencial del síndrome de Wells incluye, por supuesto, la celulitis bacteriana, pero también puede simular urticarias, eccemas, picaduras de insecto, penfigoide ampolloso o toxicodermias entre otros. El diagnóstico diferencial puede ser mucho más amplio si tenemos en cuenta todas las variedades clínicas que se han descrito de esta entidad62, 63.
Hallazgos histopatológicos
El estudio histopatológico demuestra, en la primera fase o celulítica, un edema en la dermis que está infiltrada por leucocitos, predominantemente eosinófilos, que raramente pueden extenderse a la hipodermis o al músculo. No hay signos de vasculitis. Más tarde, el infiltrado descrito se acompaña de histiocitos y aparecen las características «figuras en llama» compuestas por haces de colágeno degenerado marcadamente eosinófilo rodeadas por un infiltrado granulomatoso (fig. 7). La aparición de las «figuras en llama» se debe al depósito de la proteína básica mayor del eosinófilo en las fibras de colágeno. Aunque se pretendió que estas figuras eran patognomónicas del síndrome de Wells, se han encontrado en otras enfermedades, incluyendo las picaduras de insecto, el penfigoide ampolloso o las parasitosis, entre otras64-67. En la fase de resolución se produce una desaparición gradual de los eosinófilos, con la presencia aún de histiocitos y células gigantes alrededor de las figuras en llama formando microgranulomas62.
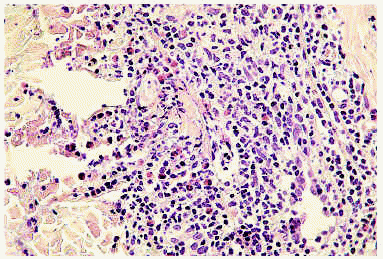
Fig. 7.--Celulitis eosinofílica: figuras «en llama».
Etiopatogenia
Aunque la patogenia de la enfermedad es desconocida, muchos la consideran resultado de una respuesta anómala del eosinófilo ante una serie de factores desencadenantes. Como tales factores desencadenantes se han descrito picaduras de insecto65, procesos mieloproliferativos67, leucemias, parasitosis y medicamentos68 entre otros. De hecho, para algunos autores el síndrome de Wells no constituye una entidad clínica distintiva y lo consideran más bien un patrón de reacción histopatológico común a muchas entidades con eosinofilia tisular65. Probablemente deba reservarse este diagnóstico para los casos con los rasgos clínicos típicos, el patrón histopatológico característico y el curso clínico recurrente.
Pronóstico y tratamiento
De los tratamientos empleados, los corticoides orales parecen los más efectivos, aunque existe un riesgo de generar corticodependencia. Otros tratamientos ensayados incluyen la dapsona, los antihistamínicos y los corticoides tópicos, pero su eficacia es difícil de evaluar debido al carácter autoinvolutivo de la enfermedad.
HIPERPLASIA ANGIOLINFOIDE CON EOSINOFILIA
La hiperplasia angiolinfoide con eosinofilia (HALE) es una enfermedad poco frecuente, descrita por primera vez por Wells y Whimster en 196969. Se trata de una entidad que Requena et al70 incluyen en el grupo de las hiperplasias vasculares cutáneas. Se ha descrito en la literatura con otras denominaciones, tales como granuloma piogénico atípico71, angioplasia papular72, nódulo inflamatorio angiomatoso73, proliferación vascular atípica intravenosa74, hemangioma histiocitoide75 y hemangioma epiteloide76. La denominación HALE es la más reconocida en la literatura ya que describe adecuadamente la enfermedad desde el punto de vista histopatológico77.
Manifestaciones clínicas
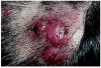
Desde el punto de vista clínico, la HALE se manifiesta como lesiones papulosas o nodulares, únicas o múltiples, agrupadas, eritematosas y de aspecto angiomatoso (fig. 8)78. En una serie amplia de 116 pacientes79, los síntomas asociados más frecuentes fueron: picor, dolor, sangrado y pulsaciones. Existe unpredominio femenino y las localizaciones más frecuentes son cabeza y cuello78. La presencia de adenopatías periféricas, así como eosinofilia y aumento de la IgE en sangre periférica son datos poco frecuentes, siendo, por el contrario, casi constantes en la enfermedad de Kimura80, entidad con la que la HALE se había identificado durante mucho tiempo81-84.
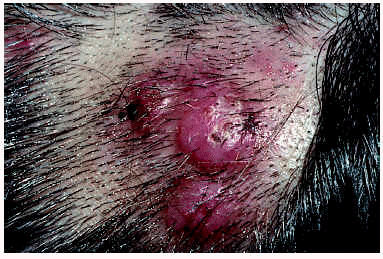
Fig. 8.--Hiperplasia angiolinfoide con eosinofilia: nódulos en cuero cabelludo.
Hallazgos histopatológicos
El estudio histopatológico se caracteriza por el hallazgo en la dermis y tejido celular subcutáneo de numerosas luces vasculares y un infiltrado linfohistiocitario con abundantes eosinófilos. Las luces vasculares están tapizadas por células endoteliales tumefactas que protruyen hacia la luz con un típico aspecto «en tachuela» (fig. 9) y adoptan, en ocasiones, un aspecto epiteloide o histiocitoide80, 81, 83. El componente inflamatorio que rodea los vasos está constituido por eosinófilos y linfocitos monomorfos que sólo en un 10% de los casos constituyen folículos linfoides.
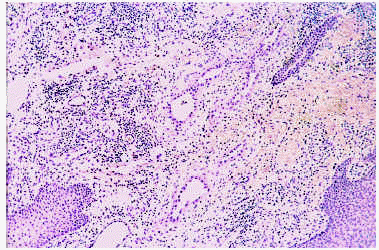
Fig. 9.--Hiperplasia angiolinfoide con eosinofilia: aspecto histológico (endotelios «en tachuela»).
Inmuhistoquímicamente las células endoteliales son positivas para el factor VIII y la lecitina Ulex europaeus-1 y son negativas para lisozima. Ultraestructuralmente se han comprobado cuerpos de Weil-Palade que confirman la estirpe endotelial de estas células77.
Etiopatogenia
Se desconoce la etiología de la HALE. Se han propuesto múltiples hipótesis, como: picaduras por insectos o parásitos69, causas infecciosas, mecanismos inmunológicos85 o estados de hiperestrogenismo en relación con la toma de anticonceptivos orales y el embarazo86.
Una teoría interesante vincula la HALE con malformaciones y shunts arteriovenosos (AV) subyacentes, en ocasiones de aparición postraumática. Olsen y Helwing79 encontraron shunt AV en el 42% de sus pacientes y esta asociación ha sido descrita posteriormente por otros autores86-89. Según esta hipótesis, la isquemia local, originada por el secuestro sanguíneo de la malformación AV, pondría en marcha la cascada renina-angiotensina. De hecho, en pacientes con HALE se ha demostrado la presencia de gránulos de renina en el citoplasma de células que rodean a los vasos88, 89.
Pronóstico y tratamiento
La HALE es una patología benigna, pero su tratamiento es difícil; se muestra resistente a múltiples terapéuticas y las recidivas son un hecho habitual. Entre las distintas modalidades terapéuticas descritas en la literatura se incluyen: corticoides tópicos, intralesionales y orales90, pentoxifilina91, indometacina92, curetaje y electrodesecación93, criocirugía94, radioterapia69, retinoides orales95, suspensión de terapia estrogénica86 y distintas modalidades de láseres96-100. La cirugía amplia y profunda sería el tratamiento de elección para la mayor parte de los autores. Se han publicado trabajos recientes a propósito de buenos resultados con el uso intralesional de interferón alfa 2a101 e interferón alfa 2b102, quizá útiles en el tratamiento de lesiones múltiples o de aquellos elementos en que la cirugía presente riesgo de resultados inestéticos.
GRANULOMA FACIAL
El GF, también denominado granuloma facial eosinofílico o granuloma facial con eosinofilia, fue descrito por primera vez en 1945 por Wigley103. Su nombre puede llevar a confundir nosológicamente esta entidad con una forma localizada de histiocitosis X llamada granuloma eosinófilo de la piel, con la que inicialmente se agrupó. Fue Lever, en 1950, el primero en establecer la distinción entre estas dos entidades104, de las que hoy sabemos que, evidentemente, no tienen ninguna relación entre sí.
El GF es considerado actualmente como una variedad de vasculitis leucocitoclástica localizada, crónica y benigna, no asociada a enfermedad sistémica, y en la que predominan los eosinófilos105.
Características clínicas
La frecuencia del GF es baja y tiene predilección por afectar a hombres de edad media y raza blanca106. Clínicamente se caracteriza por la presencia de una o, con menos frecuencia, varias pápulas o placas eritematosas bien delimitadas, de color rojo-pardo o violáceo, de tamaño variable y que en ocasiones pueden presentar telangiectasias y descamación en superficie. Los orificios foliculares son prominentes, lo cual provoca aspecto de «piel de naranja» en la superficie. Las lesiones pueden ser blandas o estar algo infiltradas a la palpación y no tienen tendencia a ulcerarse. Habitualmente son asintomáticas, aunque algunos pacientes refieren picor o sensación de quemazón. En el 10% de los casos pueden acompañarse de una discreta eosinofilia en sangre periférica106.
Como su nombre indica el GF afecta fundamentalmente a la cara (frente, nariz, mejillas), aunque existen casos de localización extrafacial, con afectación asimétrica de cuero cabelludo, tronco o extremidades107-109, e incluso formas diseminadas110. El granuloma facial extrafacial puede aparecer sin las lesiones típicas en la región facial108, 111, aunque esto no es lo más frecuente. Además, se han descritos varios casos de una entidad peculiar denominada «fibrosis eosinofílica angiocéntrica»112 que afecta a las mucosas nasal o laríngea y que parece representar una variante mucosa del GF, con el que en ocasiones puede aparecer asociada113.
Hallazgos histopatológicos
Los hallazgos histopatológicos son característicos y fundamentales para el diagnóstico de esta enfermedad. Se observa un infiltrado denso en la parte alta de la dermis que puede extenderse en profundidad, afectando incluso al tejido celular subcutáneo, y que está separado de la epidermis y de los anejos cutáneos por una banda estrecha de colágeno normal (zona Grenz). Este infiltrado es muy polimorfo y está constituido por eosinófilos, neutrófilos, linfocitos, histiocitos, células plasmáticas y, a veces, también por mastocitos.
El número de eosinófilos es variable; si bien frecuentemente constituye la célula predominante, en otras ocasiones su número es escaso. Se han realizado estudios de inmunohistoquímica que han puesto en evidencia la presencia extracelular de proteínas catiónicas de los gránulos de los eosinófilos114, indicando que estas células muchas veces existen, pero al estar «degranuladas» son poco evidentes con la micros copía óptica. La microscopía electrónica sí demuestra la presencia de gran cantidad de eosinófilos, muchas veces con alteraciones estructurales características115, 116.
Los neutrófilos suelen localizarse alrededor de los vasos sanguíneos, donde se observa leucocitoclastia. Los vasos están dilatados y suelen presentar células endoteliales prominentes. Con frecuencia los signos de vasculitis resultan evidentes, con depósito de material fibrinoide eosinófilo alrededor de los vasos. Es habitual encontrar extravasación de hematíes y depósitos de hemosiderina en la dermis, lo que explica el color pardusco de las lesiones. En este sentido, el GF se incluye, junto con el eritema elevatum et diutinum, dentro del grupo de las vasculitis leucitoclásticas crónicas y localizadas105, 117, 118, y es aún motivo de controversia el hecho de que para algunos autores estas dos enfermedades sean diferentes manifestaciones de una misma entidad119. En el infiltrado del eritema elevatum predominan los neutrófilos, los eosinófilos son escasos y no suele respetar de forma tan clara una zona libre en la dermis superior. La presencia de ésteres de colesterol en el interior de los histiocitos espumosos, mal denominada colesterolosis extracelular y típica del eritema elevatum, también puede observarse en el granuloma facial. Cuando las lesiones se cronifican, en ambas enfermedades el infiltrado inflamatorio se sustituye por áreas de fibrosis del colágeno que tiende a adoptar una disposición concéntrica o estoriforme alrededor de las paredes vasculares; este patrón histológico se ha denominado «venulitis crónica fibrosante localizada» y en él también se podrían incluir algunos de los denominados pseudotumores inflamatorios, ricos en células plasmáticas120.
Los estudios de inmunofluorescencia son poco concluyentes. Se han observado depósitos de IgG, IgA e IgM en la membrana basal de la epidermis y alrededor de las paredes vasculares, donde pueden observarse también depósitos de fibrina117.
Etiopatogenia
La etiología es desconocida. Se ha involucrado directamente al eosinófilo en la patogenia de esta enfermedad y se ha propuesto la hipótesis de una vasculitis leucitoclástica crónica inducida por un fenómeno inmunoalérgico similar a la reacción de Arthus y desencadenado por un antígeno desconocido. La reacción antígeno-anticuerpo daría lugar al depósito de inmunoglobulinas y complemento que actuarían como factores quimiotácticos para neutrófilos y eosinófilos117. La liberación de proteínas tóxicas de los gránulos de los eosinófilos desencadenaría la respuesta inflamatoria114. También se ha sugerido el posible papel de la exposición solar, ya que las lesiones casi siempre aparecen en zonas fotoexpuestas y en pacientes de raza blanca.
Pronóstico y tratamiento
Se trata de un cuadro benigno que no se asocia a enfermedades sistémicas. Sin embargo, las lesiones del GF son de curso crónico y pueden crecer de forma progresiva hasta tener varios centímetros de tamaño. Sólo excepcionalmente regresan de forma espontánea.
Suelen ser resistentes a la mayor parte de los tratamientos, con gran tendencia a la recidiva, pero quizás la crioterapia y los corticoides intralesionales deban considerarse como las primeras alternativas terapéuticas121. La extirpación quirúrgica puede ser una alternativa, pero las lesiones tienen tendencia a recurrir. Se han comunicado recientemente resultados satisfactorios con el láser de colorante pulsado122. Igualmente se ha utilizado en su tratamiento la dapsona123, 124, así como también los antipalúdicos, la colchicina y clofacimina125, con resultados variables106.
DERMATOSIS EOSINOFILICAS MUCOCUTANEAS AUTOINVOLUTIVAS
Dentro de este apartado se revisan, de manera conjunta, tres entidades: la úlcera eosinofílica de la mucosa oral, proceso bien conocido y referido con cierta frecuencia en la literatura, y otras dos patologías excepcionalmente mencionadas: el granuloma eosinofílico pseudotumoral de Kuske y la nodulosis eosinofílica transitoria. Estas tres patologías tienen en común el carácter clínico-evolutivo benigno y autorresolutivo de las lesiones, además del predominio histológico del eosinófilo entre las células que componen el infiltrado inflamatorio hallado en la dermis.
Úlcera eosinofílica de la mucosa oral
La UE es una lesión poco frecuente que se localiza generalmente en la lengua y que debería estar presente siempre entre los diagnósticos diferenciales de las lesiones ulcerosas de la mucosa oral. La descripción clínica inicial fue realizada por Riga en 1881, e histológicamente fue caracterizada por Fede en 1890. Se ha publicado en la literatura con distintos nombres, como enfermedad de Riga-Fede en niños126, granuloma traumático de la lengua127 y granuloma eosinofílico de la lengua128 entre otros.
Manifestaciones clínicas
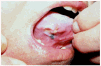
La incidencia de la UE se distribuye por igual en ambos sexos129 o es ligeramente más frecuente en mujeres según alguna serie130. Se diagnostica a cualquier edad, aunque hay un pico de incidencia entre la cuarta y sexta décadas de la vida131-133. La localización más frecuente es la lengua (fig. 10), pero también se ha descrito en otras localizaciones como labios, mucosa bucal, encías y paladar131, 134. Generalmente es una lesión única, pero hay casos múltiples y recurrentes129, 130, 135, 136. Clínicamente se trata de una úlcera de 1 a 2 cm de diámetro con los bordes indurados, asintomática o extremadamente dolorosa135, de días o escasas semanas de evolución. Más raramente puede manifestarse como una simple induración mucosa129, 130, 137.
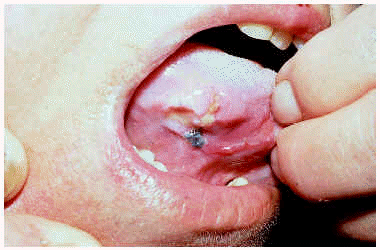
Fig. 10.--Úlcera eosinofílica de la lengua.
Hallazgos histolopatológicos
Los hallazgos del examen histológico son muy característicos. Se observa una zona ulcerada de la mucosa cubierta por una capa de exudado fibrinoide con detritus celulares (fig. 11). En la base de la úlcera hay tejido de granulación, con incremento en el número de capilares con células endoteliales prominentes. En los bordes de la úlcera el epitelio aparece hiperplásico. La submucosa está ocupada por un infiltrado difuso compuesto fundamentalmente por eosinófilos y, en cantidad variable, por linfocitos, plasmocitos e histiocitos. El infiltrado con predominio de eosinófilos llega hasta el plano muscular (fig. 12) y las fibras musculares aparecen edematosas y con alteraciones degenerativas127, 131, 134, 138. Hay autores que han descrito la presencia asociada de vasculitis leucocitoclástica138.
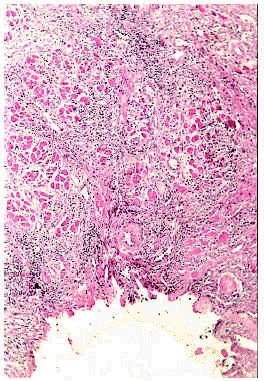
Fig. 11.--Úlcera eosinofílica: aspecto histológico. Cortesía de la doctora Emilia Fernández (Salamanca).

Fig. 12.--Úlcera eosinofílica: aspecto histológico. Cortesía de la doctora Emilia Fernández (Salamanca).
Etiopatogenia
La etiopatogenia de la UE es desconocida. El factor traumático ha sido reiteradamente considerado como la etiología de esta afección. Braskar y Lilly127 demostraron que se desarrollaba una lesión histológicamente similar en la lengua de ratas albinas mediante traumatismos repetidos. Hay autores en desacuerdo129, 139, pues en algunas series sólo se encontraron antecedentes traumáticos en el 30%-47% de los casos y no hubo claros antecedentes traumáticos en las lesiones recurrentes.
Se ha demostrado mediante tinciones inmunohistoquímicas130, 140 que el infiltrado linfocítico de la UE está compuesto predominantemente por células T y que son abundantes las células T antigenoespecíficas. Estos hallazgos sugieren que la inmunidad celular desempeña un papel importante en la patogenia de la UE. Los linfocitos T activados producen linfocinas como la interleucina-1 (IL-1) y el factor de necrosis tumoral (TNF) que favorecen la adhesión de los eosinófilos al endotelio vascular y actúan como agentes quimiotácticos para los mismos. Hay trabajos recientes que describen la UE en aparente relación con la toma de medicamentos141, 142, lo cual sugiere posibles etiologías diferentes al traumatismo local.
Pronóstico y tratamiento
Los casos publicados inicialmente fueron tratados con extirpación parcial o completa, electrocoagulación, criocirugía, infiltración con corticoides o radioterapia127, 134, 141. Sin embargo, en los últimos años se desaconseja todo tratamiento, teniendo en cuenta que la úlcera es benigna y en cuestión de semanas o meses evoluciona hacia la resolución de manera espontánea.
Granuloma eosinofílico pseudotumoral de la piel de Kuske
Esta lesión fue descrita por Kuske en 1952143 en un paciente adulto con una tumoración de aspecto peculiar en el dorso de la mano, cuya etiología no pudo determinarse y que se resolvió espontáneamente. El estudio microscópico de la biopsia demostró un infiltrado denso en la dermis con histiocitos, linfocitos y abundantes eosinófilos entre haces engrosados de colágeno. Kuske denominó esta lesión, de forma descriptiva, como granuloma eosinofílico pseudotumoral de la piel.
En la búsqueda bibliográfica sólo hemos encontrado otro trabajo reciente en el que se describe la misma entidad144. Se trata de un paciente adulto con dos lesiones tumorales que se resolvieron espontáneamente en varias semanas y con un estudio histológico superponible al definido. Los autores de este último trabajo144 proponen una serie de criterios clínicos (tumor cutáneo firme sugerente de malignidad, regresión espontánea en meses, no precedido de enfermedad infecciosa) e histológicos (infiltrados dérmicos superficiales y profundos constituidos por eosinófilos, neutrófilos, e histiocitos epitelioides y espumosos, focos de necrosis con polvillo nuclear y tinciones negativas para bacterias, micobacterias y hongos) para el diagnóstico de esta entidad.
Nodulosis eosinofílica transitoria
La nodulosis eosinofílica transitoria (NET) ha sido referida en la literatura francesa en sólo dos trabajos145, 146. Se ha descrito en edades infantiles y consiste en la aparición de múltiples nódulos en distintas partes del cuerpo, acompañados de pequeñas adenopatías, sin otra sintomatología acompañante. En un ejemplo146 las lesiones cutáneas estaban precedidas de un proceso infeccioso de vías respiratorias. Las lesiones se resolvieron espontáneamente en el curso de tres semanas. El estudio histológico mostró un infiltrado inflamatorio localizado en la dermis con predominio de eosinófilos145, en lo esencial superponible a la lesión descrita por Kuske143.
Por tanto, estas tres entidades (UE, pseudotumor de Kuske y NET) tienen en común muchos aspectos: una clínica similar, con lesiones nodulares únicas (UE) o múltiples (NET), que en el caso de la UE tienden a la ulceración, una evolución clínica benigna con tendencia a la resolución espontánea en el curso de semanas, y una etiopatogenia aún por determinar. Los hallazgos histológicos en las tres entidades son también similares: infiltrados densos dérmicos con predominio de eosinófilos. Todas estas similitudes llevan a interpretar que, posiblemente, se trata de distintos aspectos clínicos dentro del espectro de una misma enfermedad144, es decir, manifestaciones mucocutáneas reactivas similares ante distintos estímulos, como traumatismos o antígenos infecciosos o tumorales. No obstante, resulta atrevido sacar conclusiones, dado el escaso número de publicaciones acerca de unas patologías de las que, por tanto, quedan muchas incógnitas por resolver.
PAPULOERITRODERMIA
En 1984, Ofuji147 delimitó magistralmente un nuevo cuadro clínico al que denominó papuloeritrodermia (PE). Publicaciones posteriores han confirmado la individualidad clínica de un proceso cuya observación no resulta excepcional148.
Características clínicas
El diagnóstico de PE se establece, esencialmente, en función de criterios clínicos. Se trata de pacientes en edades medias o avanzadas de la vida, preferentemente hombres, con una dermatosis pruriginosa de evolución crónica. La erupción está constituida por pápulas liquenoides, aplanadas, de contorno cuadrangular, color rojo apagado a marrón, acompañadas por otras áreas más difusas, de aspecto eritrodérmico, resultantes de la confluencia de las zonas papulosas. El proceso afecta a la mayor parte de la superficie cutánea a excepción de la cara. La característica macroscópica que constituye el auténtico signo clínico definitorio de la enfermedad reside en el peculiar patrón de distribución de la erupción: la dermatosis respeta las zonas de presión como axilas, ingles, flexuras antecubital y poplítea, así como los pequeños pliegues de flexión posicional en la piel abdominal (fig. 13), lo cual se ha referido con el descriptivo término de «signo de la tumbona» (deck chair sign)149, 150.
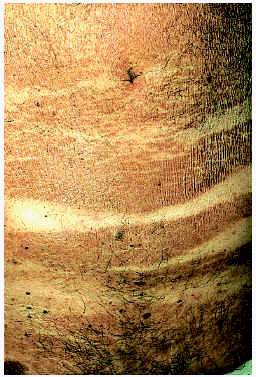
Fig. 13.--Papuloeritrodermia: «signo de la tumbona».
Hallazgos histopatológicos
Los hallazgos histológicos resultan poco característicos. Se observa, en la dermis papilar y media, la existencia de infiltrados linfocitarios perivasculares con abundantes eosinófilos. Es posible algún grado de exocitosis linfocitaria hacia la epidermis y cierto pleomorfismo celular en los infiltrados linfocitarios dérmicos151.
Etiopatogenia
El lugar nosológico que, definitivamente, habrá de ocupar la PE aún está por definir. Algunos autores han interpretado este proceso como una reacción de hipersensibilidad hacia algún antígeno no identificado151-153. El respeto de las áreas de presión y los pliegues cutáneos resultaría de una oclusión vascular funcional que impediría, localmente, la expresión clínica del brazo eferente de la respuesta inmune152. Se ha especulado con que la PE pudiera representar una variante infrecuente de dermatitis atópica en sujetos ancianos152. Por otra parte, la ocasional semejanza clínica con el síndrome de Löffler154, o la existencia de eosinofilia tisular (piel, médula ósea) y aumento de la IgE sérica155-158, han justificado que la PE se haya contemplado, en alguna ocasión, como expresión clínica peculiar de un síndrome hipereosinofílico159. Se han referido ejemplos aislados de asociación, quizá sin relación patogénica alguna, de PE con síndrome de inmunodeficiencia adquirida160, tiña del cuerpo161 o dermatitis de contacto a perfumes162.
En algunos trabajos154, 163 se describe la evolución en el tiempo de la PE hacia un linfoma T periférico primario cutáneo. Éste podría ser el resultado final bien de la complicación tardía de una dermatosis primitivamente inflamatoria, bien de la evolución de una erupción de carácter prelinfomatoso desde el comienzo.
Existen trabajos en los que se recoge la asociación de PE con algún tipo de malignidad, visceral o hematológica. Así se ha relacionado con linfoma B, linfoma T, cáncer gástrico, cáncer de pulmón164 o leucemia mielomonocítica crónica156. El escaso número de referencias y lo reciente de su descripción no permiten asegurar si la relación de la dermatosis con la enfermedad maligna interna cumple los requisitos que se han propuesto para definir un auténtico síndrome cutáneo paraneoplásico165.
Pronóstico y tratamiento
La aparente variedad de patologías subyacentes a la PE parece favorecer la hipótesis de que se trata de un patrón de expresión clínica (un síndrome) que pueden adoptar diferentes enfermedades inflamatorias o linfoproliferativas en la piel166, 167. La naturaleza de la PE no sería más específica que la de las formas clínicas habituales de eritrodermia y, como éstas, podría ser manifestación de cuadros de hipersensibilidad, erupciones prelinfomatosas o malignidad interna168. Por tanto, tras un diagnóstico de PE, los pacientes deberán someterse a procedimientos de diagnóstico y tratamiento análogos a los recomendados en otros tipos clínicos de eritrodermia169, 170.
DERMATITIS EOSINOFILICA PAQUIDÉRMICA
Sobre esta rara enfermedad, descrita por Jacyk en 1996, sólo existe una referencia en la literatura médica171. En ella se recoge la observación de tres mujeres sudafricanas jóvenes, con una erupción generalizada de pápulas pruriginosas en el seno de una piel con un engrosamiento semejante al de una dermatitis atópica intensa. Presentaban, además, hipertrofia genital marcada y una eosinofilia intensa y persistente en sangre periférica. Desde el punto de vista histológico se encontró un infiltrado inflamatorio dérmico con predominio de eosinófilos, fibrosis dérmica y cierto grado de proliferación vascular. Todos los casos respondieron favorablemente al tratamiento con dapsona y prednisolona.


