


INTRODUCCION
La condrodisplasia puntiforme (punctata) (CDP) es un defecto óseo consistente en la aparición de focos de calcificación epifisarios durante la infancia, que puede asociar además alteraciones en la piel, cara, ojos y aparato locomotor1. Se conocen cuatro tipos de CDP: la forma autosómica recesiva (CDP rizomélica o CDPR), debida a una alteración peroxisomal, la forma autosómica dominante, la forma recesiva ligada al X (CDPX-1), causada por mutaciones en el gen de la arilsulfatasa E2 y la forma dominante ligada al X (CDPX-2) o síndrome de Conradi-Hünermann-Happle o simplemente síndrome de Happle, en la que recientemente se han encontrado mutaciones en el gen de una enzima perteneciente a la cadena de síntesis del colesterol2-6. Existen otros tipos de CDP menos frecuentes, tanto hereditarios (tipos tibiometacarpiano o braquitelefalángico)1,4, como adquiridos (embriopatía por warfarina)1,4. Sin embargo, la CDP también puede aparecer como una alteración aislada en otros síndromes como el de Zellweger, el de Down, el de la trisomía 18 o en el síndrome CHILD1,7.
DESCRIPCION DEL CASO
Una niña, nacida por cesárea por presentación podálica tras 37 semanas de gestación, de madre primigesta, presentó al nacer un peso de 2.370 g (percentil = 25) y una talla de 36 cm (percentil < 3). La cesárea transcurrió sin incidencias. El desarrollo del embarazo fue normal, pero en las ecografías sistemáticas realizadas se observó una disminución de las medidas fetales, realizándose un diagnóstico de «posible acondroplasia». Su abuela materna había fallecido a causa de una esclerosis lateral amiotrófica y su padre presentaba pequeñas opacidades corneales desde hacía 5 años, que se encontraban en estudio, sin filiar. Los padres no eran consanguíneos y la madre estaba sana. En el momento del nacimiento la niña presentaba buen estado general y vitalidad, con cuello corto y lateralizado, abdomen asimétrico, miembros inferiores cortos, pies zambos y rotación externa a 180º del pie derecho, junto con escoliosis. Toda la piel estaba eritematosa y mostraba una importante hiperqueratosis y descamación. El resto de la exploración clínica fue normal.
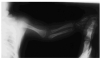
Una serie ósea mostró una displasia espondilopoliepifisaria con punteado de las epífisis de los huesos largos (fig. 1). La radiografía de tórax y las ecografías de abdomen y cerebro fueron por lo demás normales. Las determinaciones analíticas de sangre y orina no mostraron resultados anormales. Se practicó una biopsia cutánea, que mostró una intensa hiperqueratosis ortoqueratósica, con granulosa normal, dilatación comedoniana de los infundíbulos pilosos y focos de calcificación de queratina. El cariotipo fue 46XX.
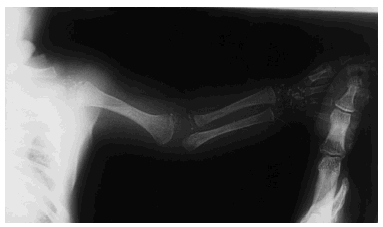
Fig. 1.--Punteado de las epífisis de los huesos de la extremidad superior.
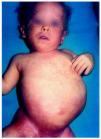
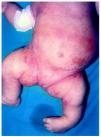
Al mes y medio de vida fue remitida a nuestro servicio para valoración. En ese momento presentaba un eritema difuso, con escamas nacaradas, secas y adheridas, en zonas frontalinterciliar, preauriculares, en cuero cabelludo y dorso de pies (fig. 2). En la zona de pliegues del pañal presentaba áreas eritematosas descamativas (fig. 3). También presentaba áreas de alopecia no cicatrizal en cuero cabelludo y moderada hipertricosis en ambos miembros inferiores. La paciente fue tratada con cremas emolientes y corticoides tópicos, con mejorías parciales y brotes periódicos. A los 3 meses de edad podía apreciarse la malposición intensa de ambas extremidades inferiores, con acortamiento de éstas y limitación de la movilidad de ambas caderas. La nariz era corta y evertida, con hundimiento de la raíz nasal. El tono muscular estaba muy disminuido en tronco y extremidades. En la exploración oftalmológica presentaba pequeñas calcificaciones en ambos ojos, más importantes en el izquierdo. Se realizó una ecografía abdominal, en la que se visualizaran riñones malformados, pelvis renal derecha extrarrenal y ampulosa y dilatación del uréter distal.
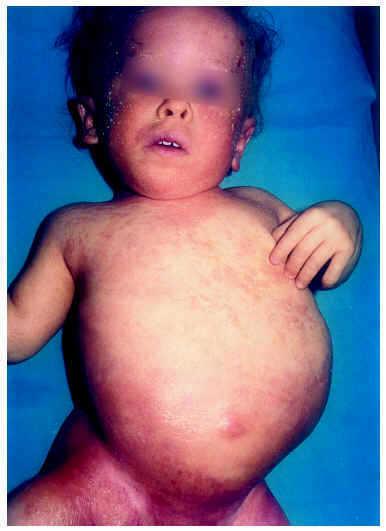
Fig. 2.--Eritema difuso y descamativo en cabeza y tronco.
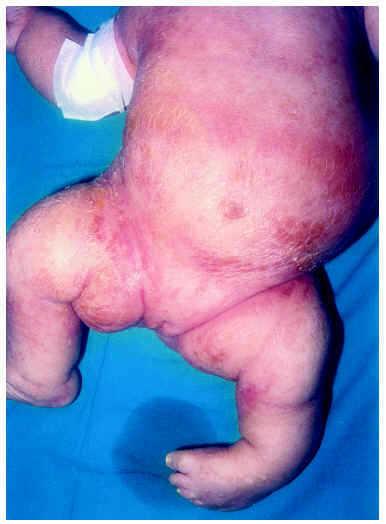
Fig. 3.--Eritema y descamación en tronco y miembros inferiores. Malposición grave de ambos miembros.
Se realizaron mediciones en sangre de ácido fitánico, plasmalógeno y ácidos grasos de cadena muy larga, y en orina de oligosacáridos y glucosaminoglucanos, que resultaron normales. En fibroblastos cultivados se midieron las concentraciones de enzimas de betaoxidación (oxidasa y tiolasa), dihidroxiacetona fosfato aciltransferasa (DHAPT) y catalasa intracelular, biosíntesis de novo de plasmalógeno y fraccionamiento de ácidos grasos de cadena muy larga, sin encontrar ninguna alteración. Estos resultados descartaron una alteración peroxisomal causante de una CDP rizomélica.

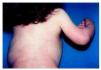
Fig. 4.--Lesiones eritematosas y descamativas siguiendo un patrón lineal de líneas de Blaschko en el tronco.
A lo largo de la evolución de la paciente se produjo una mejoría de las lesiones cutáneas, que comenzaron a tomar un aspecto lineal en el tronco y miembros (fig. 4) y posteriormente, el eritema con descamación dio paso a una atrofodermia puntiforme. La paciente también desarrolló cataratas sectoriales bilaterales, más marcadas en ojo izquierdo. Actualmente tiene 6 años, y presenta todavía graves malposiciones óseas que le impiden la deambulación.
DISCUSION
En 1914, Conradi realizó la primera descripción de este síndrome sobre un caso de «condrodisplasia fetal hipoplásica»7. En 1931, Hünermann aportó otro caso similar y Spranger et al pusieron de manifiesto en 1971 la heterogeneidad genética de la CDP y distinguieron dos tipos: dominante y recesiva7. Posteriormente se añadieron las formas recesiva y dominante ligadas al cromosoma X. Entre 1977 y 1979, Happle3,7,8 describió definitivamente la forma dominante ligada a X.
La CDP dominante ligada al cromosoma X se manifiesta sólo en mujeres, ya que para los fetos varones es potencialmente letal4,7. Sin embargo, existen casos publicados de varones con CDPX-29,10. Los fetos del sexo femenino continúan su desarrollo gracias al fenómeno de lionización, que inactiva uno de los dos cromosomas X al azar. Seguramente este fenómeno sea el responsable de la asimetría de las manifestaciones clínicas, así como de la distribución de las lesiones cutáneas o de la diferente expresividad clínica entre los pacientes2-7.
Al nacimiento se observa una eritrodermia ictiosiforme que a los pocos meses de vida evoluciona a una ictiosis lineal que sigue las líneas de Blaschko, y se acompaña de hiperqueratosis palmoplantar, alopecia cicatrizal, atrofodermia folicular, hiperpigmentación estriada y distrofias ungueales y dentales. El componente ictiosiforme suele mejorar en la adolescencia aunque a veces puede persistir en la edad adulta, mientras que la alopecia cicatrizal y la atrofodermia folicular persisten de por vida. Además, se asocia otra serie de alteraciones no cutáneas, cuya principal característica es la distribución asimétrica. Dentro de las alteraciones musculoesqueléticas se encuentran estatura baja, acortamiento asimétrico de los miembros, escoliosis con alteraciones vertebrales, frente aplanada, puente nasal plano, micrognatia, alteraciones del paladar, deformidades craneales, sindactilia, pie equino, luxación congénita de cadera y contracturas en flexión de codo y rodilla7,8. También presentan alteraciones oculares como cataratas, por lo general unilaterales.
Los hallazgos histopatológicos muestran una intensa hiperqueratosis, con pigmentación de la basal y tapones de queratina en los folículos pilosos, que contienen depósitos de calcio11. En las radiografías óseas se observan depósitos de calcio en los cartílagos epifisarios.
Generalmente, las mutaciones están producidas de novo, aunque se han descrito casos familiares7,9; en estas ocasiones se produce el fenómeno de anticipación. Este fenómeno, consistente en un aumento de la expresividad clínica de una enfermedad de generación en generación, es explicado por la presencia de mutaciones inestables en tripletes de bases repetidas; en el caso de la CDPX-2 el fenómeno de anticipación se explicaría por la presencia de un mosaicismo somático y/o gonadal, por el que se afectan sólo ciertas líneas celulares, permitiendo la coexistencia de madres asintomáticas o paucisintomáticas, con hijas muy afectadas y a edades más tempranas. También se ha querido implicar la inactivación al azar del cromosoma X para explicar el fenómeno de anticipación3,9. Se han realizado múltiples estudios para determinar las alteraciones bioquímicas causantes de este síndrome; a pesar de que varios autores han asegurado que la CDPX-2 presenta alteraciones en los peroxisomas, tal como ocurre en la forma autosómica recesiva8,12-16, recientemente se han detectado alteraciones en los productos intermedios de la síntesis del colesterol2,17,18, que se han relacionado con mutaciones en el gen de la proteína ligadora de emopamil3,4-6,19-22. Esta es una enzima participante en la síntesis de colesterol, que tiene una función de Δ8-Δ7esterol isomerasa y convierte el colesterol-8(9)-3-β-ol en lanosterol18, que a su vez es convertido en 7-deshidrocolesterol, el mayor precursor de síntesis de colesterol y vitamina D2,5,6. Se han encontrado, por el momento, 15 tipos diferentes de mutaciones en el gen de la proteína ligadora de emopamil, localizado en la región Xp11.22-Xp11.23, posiblemente causantes de este síndrome: una deleción, dos alteraciones estructurales, seis mutaciones que originan codones de terminación precoz de la cadena, cuatro mutaciones de sustitución puntual de aminoácidos y dos sustituciones intrónicas3-6,23,24.


