






El carcinoma de células de Merkel (CCM) es una neoplasia neuroendocrina maligna. Con frecuencia existe diseminación ganglionar o metástasis al diagnóstico. Realizamos un estudio descriptivo retrospectivo de los pacientes con CMM del Hospital Universitario Fundación Alcorcón entre enero/1998 y diciembre/2018. En 21 años diagnosticamos 11 pacientes con CCM: 7 varones (63%) y 4 mujeres (36%), con una edad media de 77,6 años. El 45% de los pacientes presentaron un estadio IIIB (pTNM) al diagnóstico. Todos los pacientes menos uno, fueron subsidiarios de cirugía local, identificándose en 7 casos invasión linfovascular. Tras la cirugía, 5 pacientes recibieron radioterapia adyuvante y 3 quimioterapia adyuvante. El 54% fallecieron por el tumor (tiempo medio supervivencia: 14,5 meses). El CCM es una neoplasia maligna infrecuente cuya incidencia se sitúa en 0,18-0,41 casos/100.000 habitantes/año, similar a los 0,29-0,32 casos/100.000 habitantes/año registrados en nuestra serie. Recientemente ha sido aprobado avelumab para casos metastásicos con esperanzas prometedoras.
Merkel cell carcinoma (MCC) is a malignant neuroendocrine tumor. Metastasis or lymph node spread is often detected at diagnosis. We performed a descriptive, retrospective study of patients diagnosed with MMC at Hospital Universitario Fundación Alcorcón in the Community of Madrid, Spain between January 1998 and December 2018. Eleven patients (7 men [63%] and 4 women [36%]; mean age, 77.6 years) were diagnosed with MCC during this 21-year period; 45% of patients had stage IIIB disease (pTNM) at diagnosis. All patients but one underwent local surgery, and lymphovascular invasion was detected in 7 cases. Eight patients received adjuvant therapy after surgery (radiation therapy in 5 cases and chemotherapy in 3). Six patients (54%) died of MCC (mean survival, 14.5 months). MCC is an uncommon malignant tumor with an annual incidence of around 0.18 to 0.41 cases per 100 000 inhabitants; this is similar to the rate of 0.29 to 0.32 cases per 100 000 inhabitants a year detected in our series. Results with avelumab, a drug recently approved for the treatment of metastatic MCC; have been promising.
El carcinoma de células de Merkel (CCM) es una neoplasia neuroendocrina maligna infrecuente, que a menudo se presenta en estadios avanzados. Se localiza en zonas fotoexpuestas de pacientes de edad avanzada, y con frecuencia existe diseminación ganglionar o metástasis al diagnóstico. En las últimas décadas su incidencia está incrementándose debido al envejecimiento de la población, al aumento de la esperanza de vida y a una mejora en las técnicas diagnósticas (técnicas inmunohistoquímicas)1.
Existe poca información acerca de la incidencia del carcinoma de Merkel en España. Es por esto que el objetivo primario del estudio fue realizar un análisis descriptivo de los datos demográficos, clínicos e histopatológicos, describiendo la tasa de incidencia de CCM de nuestra serie. Como objetivo secundario nos planteamos describir la mediana de tiempo de supervivencia de nuestros pacientes.
Material y métodosRealizamos un estudio descriptivo retrospectivo de todos los pacientes diagnosticados de CMM en el Hospital Universitario Fundación Alcorcón (HUFA) entre enero/1998 y diciembre/2018. Hasta 2011 la población de referencia de nuestro hospital era de 273.703 habitantes, cifra reducida a 103.000 habitantes desde 2012. A través de la historia clínica electrónica y de la base de datos del servicio de anatomía patológica, recogimos todas las variables demográficas (sexo, edad al diagnóstico), clínicas (tipo de lesión, tamaño tumoral máximo, presencia de ulceración), histopatológicas (celularidad tumoral, técnicas de inmunohistoquímica, índice de proliferación celular), tratamiento (tipo de cirugía, radioterapia, quimioterapia), evolución y tiempo de supervivencia. Los datos obtenidos fueron introducidos en una base de datos anonimizada y analizados con el paquete estadístico SPSS® (version 20.0; SPSS Inc, Chicago IL, EE. UU.). Las variables cualitativas fueron expresadas en porcentaje mientras que las cuantitativas se expresaron con mediana y rango. El estudio cuenta con la aprobación del Comité Ético de Investigación con medicamentos del HUFA.
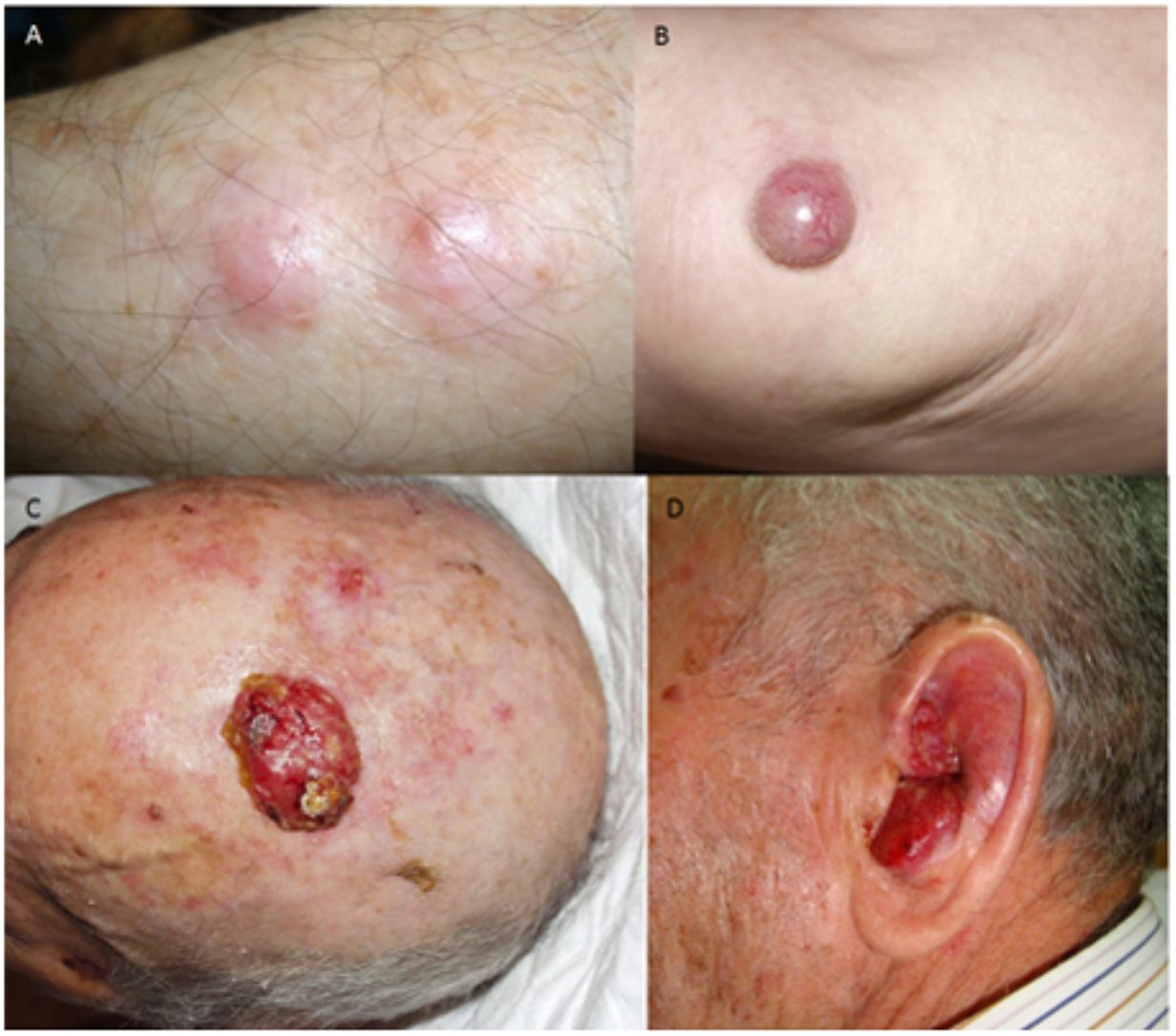
ResultadosEn 21 años (1998-2018) hemos diagnosticado 11 pacientes con CCM (tabla 1). Se trataba de 7 varones (63%) y 4 mujeres (36%), con una edad media al diagnóstico de 77,6 años y una mediana de 81 años (rango: 60-92). El 45% de los tumores se localizaron en cabeza y cuello (5/11), otro 45% en las extremidades (5/11) y solo un caso en el tronco. Todos los pacientes presentaban fototipo II-III de Fitzpatrick y ninguno inmunosupresión. Clínicamente las lesiones demostraron un gran polimorfismo. Preferentemente se presentaron como nódulos subcutáneos (fig. 1A) o tumores exofíticos eritematosos de superficie lisa (fig. 1B), asintomáticos y de rápido crecimiento. Tres casos se presentaron como tumores exofíticos de superficie erosionada (figs. 1C y D), y en un caso como placa eritematosa descamativa de bordes mal delimitados. En 7/11 pacientes (63%) el diámetro mayor del tumor primario al diagnóstico fue >2cm (cT2). Cuatro pacientes presentaron afectación linfática regional en la exploración física en el momento del diagnóstico que fue confirmada histológicamente tras punción-aspiración con aguja fina (PAAF). Además, otro paciente presentó satelitosis en el postoperatorio inmediato tras la cirugía del tumor primario (fig. 2A); por lo que el 45% (5/11) presentaban un estadio IIIB (pTNM) al diagnóstico. Solo un paciente comenzó con metástasis hepáticas, pulmonares y óseas, confirmadas histológicamente mediante autopsia tras el fallecimiento (estadio IV). En 2 pacientes realizamos biopsia selectiva de ganglio centinela (BSGC) resultando negativa en un caso y positiva en el otro. Un tercer paciente fue programado para realizar BSGC, pero su realización se desestimó por migración del radiotrazador a 4 territorios ganglionares en la linfoescintigrafía previa a la cirugía.
Datos de los pacientes
| Sexo | Edad | Localización | Al diagnóstico | PPCC | Estadio | Tratamiento | ||
|---|---|---|---|---|---|---|---|---|
| 1 | V | 84 | Brazo | 2,3cm | Adenopatías | PAAF + | pIIIB | Cx+RT |
| 2 | V | 81 | Concha auricular | 2,5cm | Adenopatías | PAAF + | pIIIB | RT |
| 3 | M | 82 | Rodilla | 2cm | Adenopatías, metástasis | PAAF + | pIV | Cx+QT |
| 4 | V | 60 | Frente | 1cm | Satelitosis | pIIIB | Cx+RT/QT | |
| 5 | V | 78 | Muslo | 0,7cm | BSGC − | pI | Cx | |
| 6 | M | 92 | Frente | 5 cm | Adenopatías | PAAF + | pIIIB | Cx+RT |
| 7 | V | 75 | Cuero cabelludo | 5cm | Pérdida de seguimiento | |||
| 8 | M | 79 | Mano | 1,5cm | Adenopatías | PAAF + | pIIIB | Cx |
| 9 | M | 81 | Cuero cabelludo | 5cm | Pérdida seguimiento | Cx | ||
| 10 | V | 81 | Pierna | 1,1cm | BSGC + | pIIIA | Cx+RT | |
| 11 | M | 61 | Flanco | 7cm | Escintig | cIIA | Cx+RT/QT |
La edad se expresa en años.
BSGC: biopsia selectiva del ganglio centinela; Cx: cirugía radical; M: mujer; PAAF: punción aspiración con aguja fina; PPCC: pruebas complementarias; QT: quimioterapia; RT: radioterapia; V: varón.
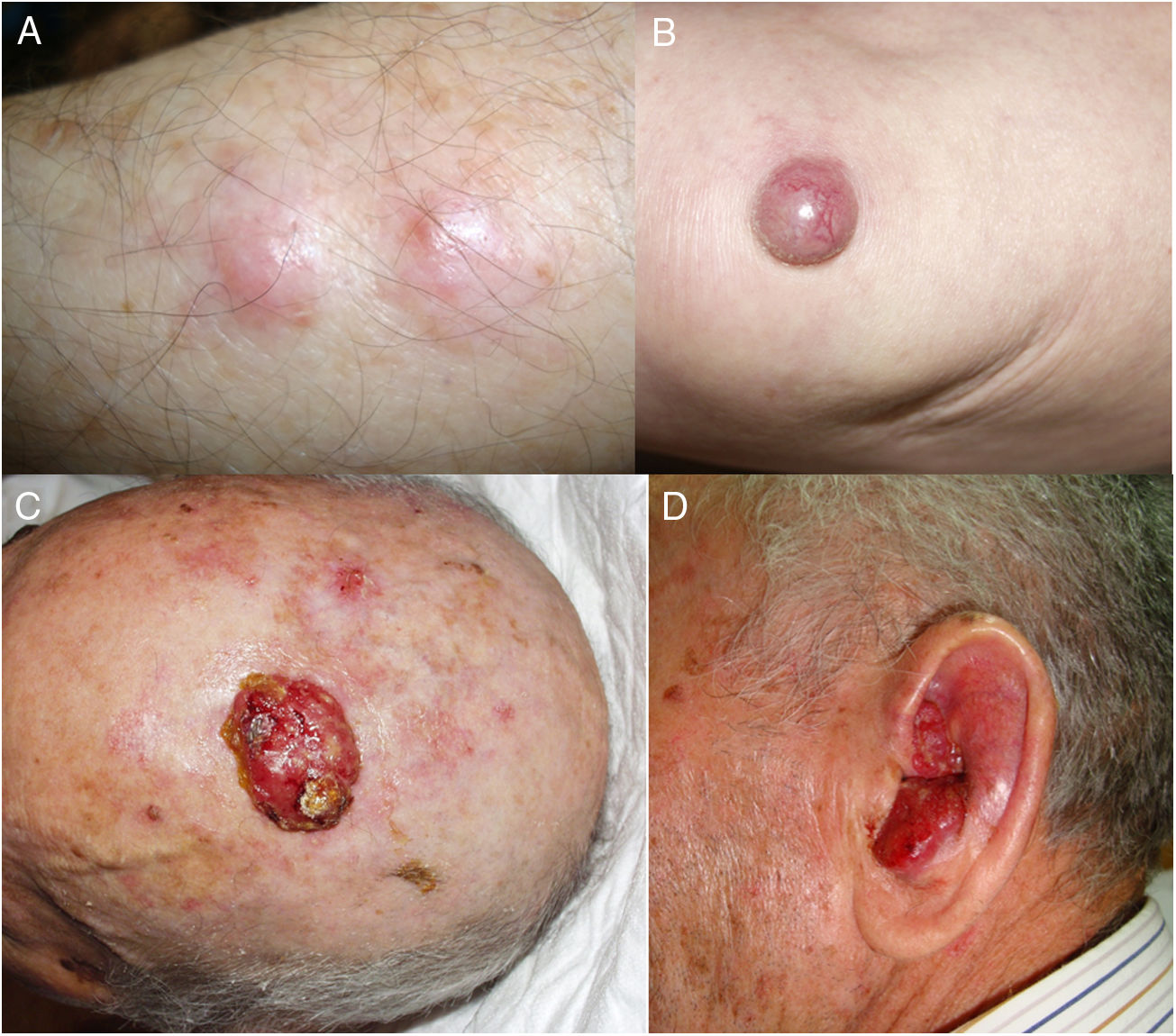
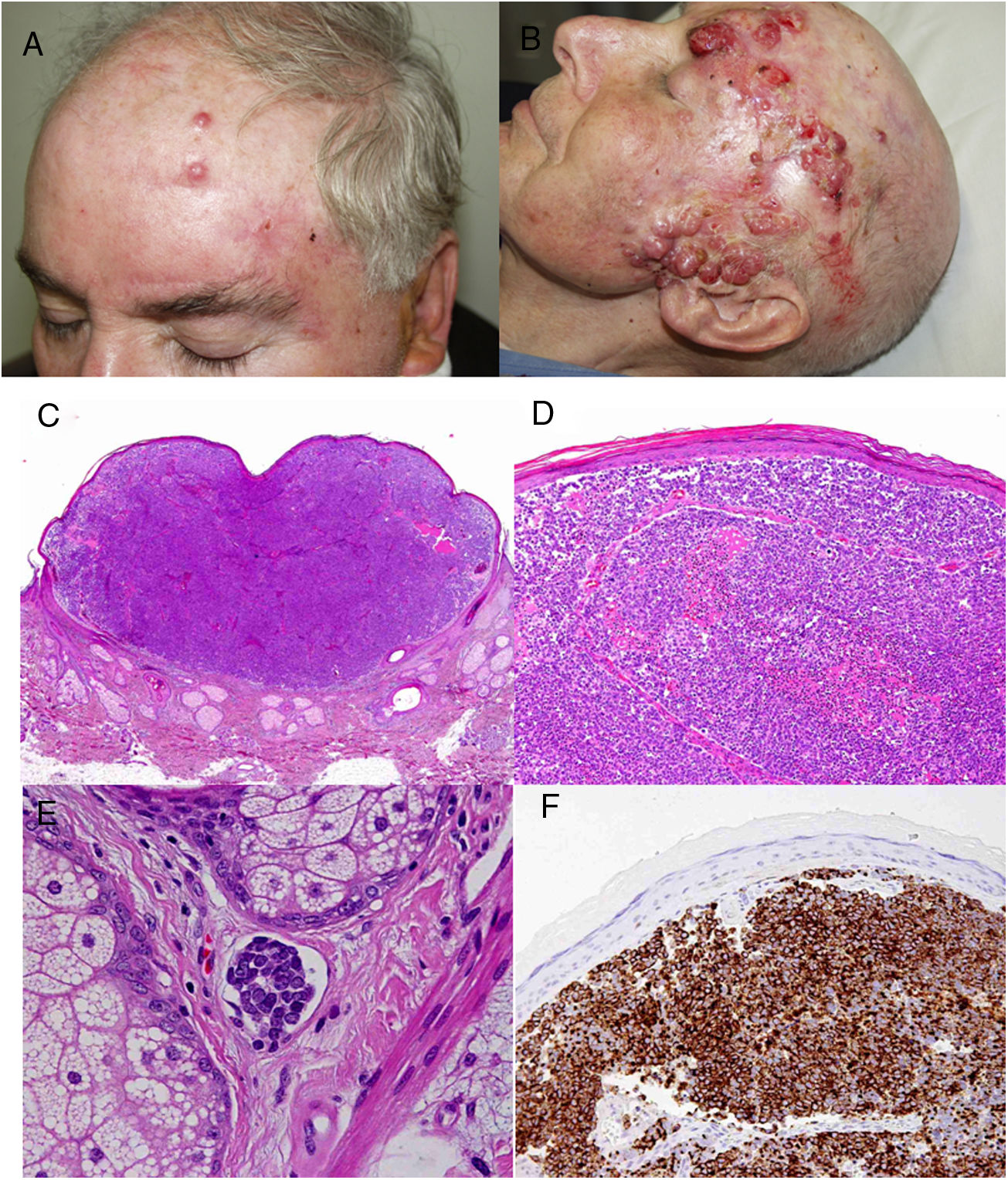
Todos los pacientes menos uno (fig. 1D), fueron subsidiarios de cirugía radical local, con márgenes clínicos de entre 1 y 3cm según la localización anatómica. El estudio histopatológico mostró en todos los casos tumoraciones de células redondas, discohesivas, de escaso citoplasma basófilo y núcleos con cromatina granular, con varios nucléolos y abundantes mitosis y apoptosis, que se disponían preferentemente en nidos sólidos (fig. 2). La inmunohistoquímica mostró positividad para CK20 en gota paranuclear y cromogranina en todos los casos (fig. 2F). CK7 y TTF1 fueron negativos. El índice de proliferación (Ki-67) resultó elevado en la mayoría de los pacientes. Siete de los 9 pacientes operados (uno inoperable, una pérdida de seguimiento tras biopsia confirmatoria), presentaron invasión linfovascular (fig. 2E).
El único paciente no candidato a cirugía recibió radioterapia (RT) local y regional neoadyuvante. Tras la cirugía, otros 5 pacientes recibieron RT adyuvante (2 RT local y 3 RT local y ganglionar) y 3 pacientes recibieron quimioterapia (QT) adyuvante; platinos y etopósido como primera línea (fig. 2B). Sin embargo, 6/11 pacientes (54%) fallecieron a causa del tumor con un tiempo medio de supervivencia de 14,5 meses y una mediana de tiempo de supervivencia de 9 meses (rango: 3-45 meses). Otros 2 pacientes fallecieron por otras causas (infección respiratoria e insuficiencia hepática) tras más de 6 años de seguimiento, mientras que 2 pacientes presentaron pérdida de seguimiento. Hasta el momento solo un paciente permanece vivo y libre de enfermedad tras 23 meses de seguimiento.
Discusión y conclusionesEl CCM es una neoplasia maligna infrecuente de curso evolutivo muy agresivo descrita por Toker en 19721. Su incidencia está aumentando, siendo Australia el país con mayor tasa con 1,6 casos/100.000 habitantes/año2,3. En EE. UU. y Europa la incidencia se sitúa en 0,18-0,41 casos/100.000 habitantes/año y se esperan 2.500 nuevos casos al año en la Unión Europea, de los cuales se estima que 1.000 de esos pacientes mueran por el tumor4–6. La incidencia calculada en nuestra serie es de 0,29-0,32 casos/100.000 habitantes/año, dato que coincide con la literatura europea. Solo hemos encontrado un estudio español que recoge la tasa de incidencia de CCM en España en una serie de 19 casos en Gerona entre 1995-2005 (1,3 casos/100.000 habitantes/año)7. El hecho de que la incidencia de nuestra serie sea menor a la descrita en la serie de Gerona puede ser debido a que nuestro estudio incluye únicamente los casos de un solo centro hospitalario mientras que la serie catalana recoge todos los casos de la población de la provincia.
El CCM es un tumor con una alta agresividad, y con frecuencia el diagnóstico se realiza en estadios avanzados. Hasta un 27-32% de los pacientes según las distintas series publicadas en la literatura presenta diseminación linfática al diagnóstico8,9. De hecho, el conocimiento del estado ganglionar es el factor pronóstico más importante, con tasas de supervivencia a los 5 años del diagnóstico de un 51% en tumores localizados y <14% en caso de enfermedad a distancia10,11. En nuestra serie, el 36,3% de los pacientes (4/11) presentaron ganglios patológicos en la exploración física realizada en el momento del diagnóstico, confirmándose en todos los casos la afectación ganglionar mediante PAAF. Aproximadamente un tercio de los pacientes con ganglios clínicamente negativos tienen enfermedad ganglionar microscópica, por esto, la BSGC constituye una herramienta de estadificación mínimamente invasiva. En nuestra serie solo pudimos plantear la realización de la BSGC a 3 pacientes debido al estadio clínico avanzado de la mayoría de los pacientes. La alta tasa de mortalidad descrita en nuestra serie creemos que está en relación con la edad avanzada (edad media: 77,6 años) y con el estadio avanzado de la enfermedad (63,6% con un estadio pIIIA o superior al diagnóstico) y, por tanto, con el alto número de pacientes con afectación ganglionar al diagnóstico.
En 2009 la American Joint Committee on Cancer (AJCC) desarrolló el primer consenso de estadificación tras analizar 5.823 casos del National Cancer Database (NCDB) que ha sido recientemente actualizado en 20178,12. Actualmente, la octava edición de estadificación propuesta por la AJCC establece una diferencia entre estadio clínico y patológico en función de la confirmación histológica de las metástasis ganglionares y/o a distancia, respectivamente (tablas 2 y 3). En nuestra serie llama la atención el avanzado estadio clínico y/o patológico que presentaron los pacientes: 7 pacientes presentaron un estadio pIIIA o superior al diagnóstico (un paciente estadio pIIIA, 5 pacientes estadio pIIIB y un paciente estadio pIV). Esto probablemente se deba al tamaño de tumor primario (5 pacientes con tumores ≤5cm) y al elevado índice de proliferación celular (Ki-67) evidenciado en el estudio histopatológico.
Clasificación clínica de la AJCC (8.ª edición)
| Estadios clínicos (cTNM) | T | N | M |
|---|---|---|---|
| 0 | Tis | N0 | M0 |
| I | T1 | N0 | M0 |
| IIA | T2-3 | N0 | M0 |
| IIB | T4 | N0 | M0 |
| III | T0-4 | N1-3 | M0 |
| IV | T0-4 | Cualquier N | M1 |
AJCC: American Joint Committe on Cancer; cN0: sin afectación ganglionar en la exploración física o mediante pruebas de imagen; cN1: afectación ganglionar clínica; cN2: metástasis en tránsito sin afectación ganglionar; cN3: metástasis en tránsito y afectación ganglionar clínica; M0: sin metástasis a distancia; M1a: metástasis a distancia cutáneas, subcutáneas o metástasis ganglionares distantes; M1b: metástasis pulmonares; M1c: otras metástasis a distancia; Tis: tumor primario in situ; T0: sin tumor primario; T1: tumor primario ≤2cm; T2: tumor primario >2 y ≤5cm; T3: tumor primario >5cm; T4: tumor primario que invade fascia, músculo, cartílago o hueso.
Clasificación patológica de la AJCC (8.ª edición)
| Estadios patológicos (pTNM) | T | N | M |
|---|---|---|---|
| 0 | Tis | N0 | M0 |
| I | T1 | N0 | M0 |
| IIA | T2-3 | N0 | M0 |
| IIB | T4 | N0 | M0 |
| IIIA | T1-4T0 | N1a (sn) o N1aN1b | M0 |
| IIIB | T1-4 | N1b-3 | M0 |
| IV | T0-4 | Cualquier N | M1 |
AJCC: American Joint Committe on Cancer; M0: sin metástasis a distancia; M1a: metástasis a distancia cutáneas, subcutáneas o metástasis ganglionares distantes; M1b: metástasis pulmonares; M1c: otras metástasis a distancia; pN0: sin afectación ganglionar en el estudio histológico; pN1a: metástasis ganglionares ocultas identificadas en la resección linfática; pN1a (sn): metástasis ganglionares ocultas identificadas en el estudio de la BSGC; pN1b: metástasis ganglionares clínicas o identificadas en pruebas de imagen y confirmadas histológicamente; pN2: metástasis en tránsito confirmadas histológicamente sin afectación ganglionar; pN3: metástasis en tránsito y ganglionares confirmadas histológicamente; Tis: tumor primario in situ; T0: sin tumor primario; T1: tumor primario ≤2cm; T2: tumor primario >2 y ≤5cm; T3: tumor primario >5cm; T4: tumor primario que invade fascia, músculo, cartílago o hueso.
No existe consenso sobre las técnicas de imagen más adecuadas a realizar como estudio de extensión en pacientes con CCM. Tanto las últimas guías americanas como las guías de práctica clínica de la Academia Española de Dermatología y Venereología (AEDV) hablan de la PET/TC de cuerpo completo como la prueba de imagen a elegir para valorar la extensión tumoral13. En nuestro caso el estudio de extensión de todos los pacientes lo llevamos a cabo con TC debido a la falta de disponibilidad de PET/TC en nuestro centro.
Si es posible, el tratamiento del CCM debe incluir una extirpación quirúrgica con márgenes de 1-2cm. A pesar de la ausencia de estudios prospectivos, la CMM no ha demostrado diferencias significativas ni en la presencia de tumor residual o en los márgenes quirúrgicos afectos ni en la supervivencia global14. Si la exploración física no evidencia afectación ganglionar, la extirpación quirúrgica debe acompañarse de una BSGC. Además, si existen factores de alto riesgo (tumor >1cm, márgenes quirúrgicos afectos o insuficientes, afectación linfovascular o localización en cabeza y cuello), las guías actuales recomiendan aplicar RT 50-66Gy sobre el lecho tumoral15. En estadios avanzados y en función de la presencia de diseminación linfática y/o metástasis a distancia, la RT del territorio ganglionar y la quimioterapia pueden ser necesarias.
Avelumab es un anticuerpo monoclonal contra el PD-L1 aprobado en marzo de 2017 por la Food and Drug Administration (FDA) y en septiembre de 2017 por la European Medicines Agency (EMA) para el CCM metastásico16. Nosotros no hemos podido probar este fármaco, ya que nuestro último paciente fue diagnosticado en febrero 2017, cuando este fármaco no tenía aprobación. Pembrolizumab y nivolumab están actualmente siendo evaluados en ensayos clínicos con resultados prometedores17,18.
En conclusión, el CCM es una neoplasia maligna muy agresiva. Con frecuencia el diagnóstico es tardío. Actualmente la aprobación de nuevos fármacos de inmunoterapia ha supuesto una esperanza para pacientes con CCM metastásico.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


