






INTRODUCCIÓN
El carcinoma de células de Merkel (CCM) fue descrito por Toker1 en 1972 como un carcinoma trabecular de la piel. Posteriormente ha recibido múltiples nombres, entre otros, merkeloma y carcinoma neuroendocrino cutáneo, este último por tener rasgos histológicos, ultraestructurales e inmunohistoquímicos similares a los tumores derivados del sistema APUD (amine and precursor uptake and decarboxilation)2 . El CCM es un tumor cutáneo poco frecuente, que se observa con mayor incidencia en individuos de raza blanca, mayores de 60 años, asociado a otras neoplasias cutáneas. La localización suele ser cabeza y cuello, principalmente periorbitaria3 , y el pronóstico, muy grave4 .
MATERIAL Y MÉTODOS
Se incluyen 5 pacientes que acudieron a nuestro hospital en los últimos 3 años por CCM. En todos ellosse realizó estudio histológico e inmunohistoquímico. Se siguió su tratamiento y evolución.
RESULTADOS
Las características clínicas y epidemiológicas se resumen en la tabla 1. Se trataba de 3 varones y 2 mujeres de entre 58 y 89 años de edad. Tres pacientes acudieron primeramente al servicio de dermatología; uno, a medicina interna, y otro, a cirugía general. Tres de ellos presentaban como antecedentes dermatológicos numerosas queratosis actínicas; uno, varios carcinomas basocelulares, y otro, enfermedad de Bowen y un carcinoma espinocelular de labio.
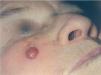
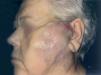
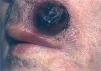
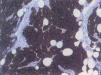
En 4 pacientes se observaba un nódulo indoloro, eritematoso, bien delimitado, de consistencia variable, cuyo tamaño oscilaba de 0,8 y 2 cm. Uno de estos 4 casos presentaba una lesión de aspecto aspecto brillante con telangiectasias (fig. 1). En el quinto se trataba de una placa eritematosa violácea de bordes mal definidos con nódulos subcutáneos de aspecto carnoso de 6 × 6 cm (fig. 2). Las localizaciones afectadas fueron mejilla (2 casos), labio superior (fig. 3), pierna y nalgas. El tiempo de evolución fue de meses. Un paciente mostraba metástasis cutáneas. En el momento del diagnóstico, otro presentaba adenopatías submandideados u ovalados de cromatina pulverulenta, sin nucléolos (fig. 4). Las técnicas inmunohistoquímicas fueron positivas para enolasa neuroespecífica (fig. 5), cromogranina, sinaptofisina y citoqueratinas AE1/AE3 (fig. 6) y negativas para antígeno leucocitario común y S-100.
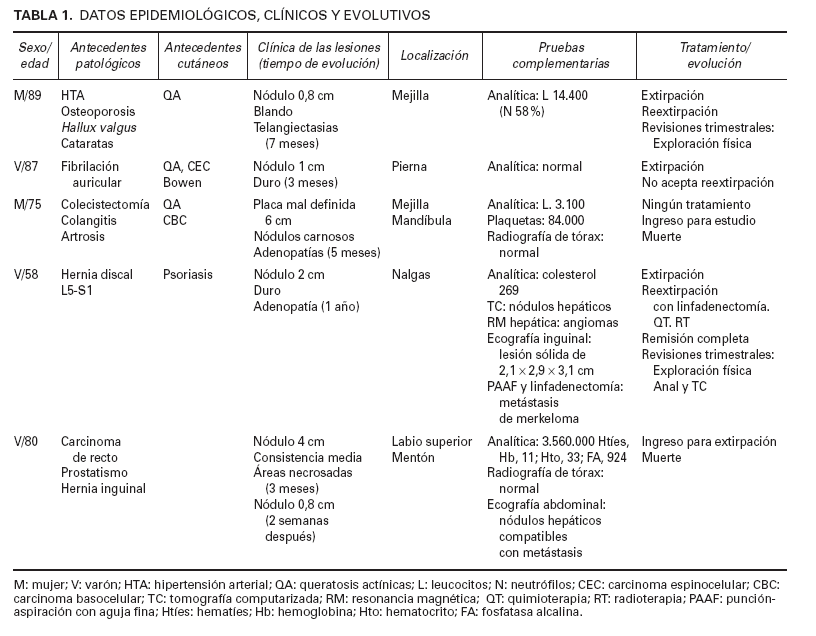
TABLA 1. DATOS EPIDEMIOLÓGICOS, CLÍNICOS Y EVOLUTIVOS
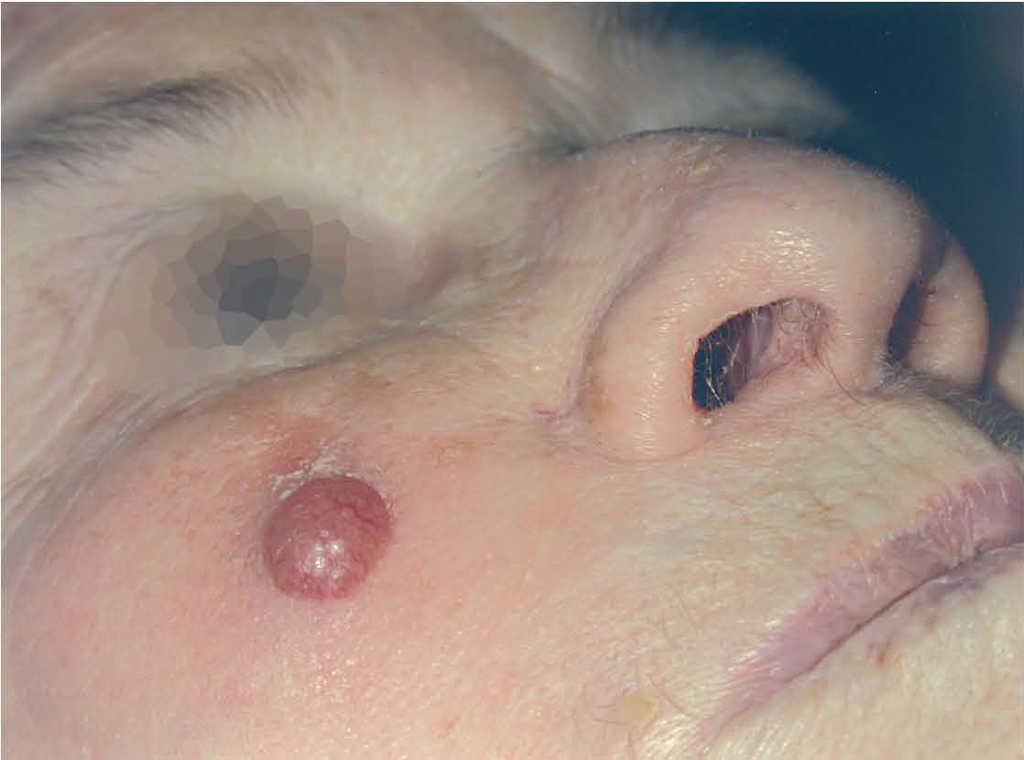
Fig. 1.—Nódulo eritematoso brillante con telangiectasias en la superficie.
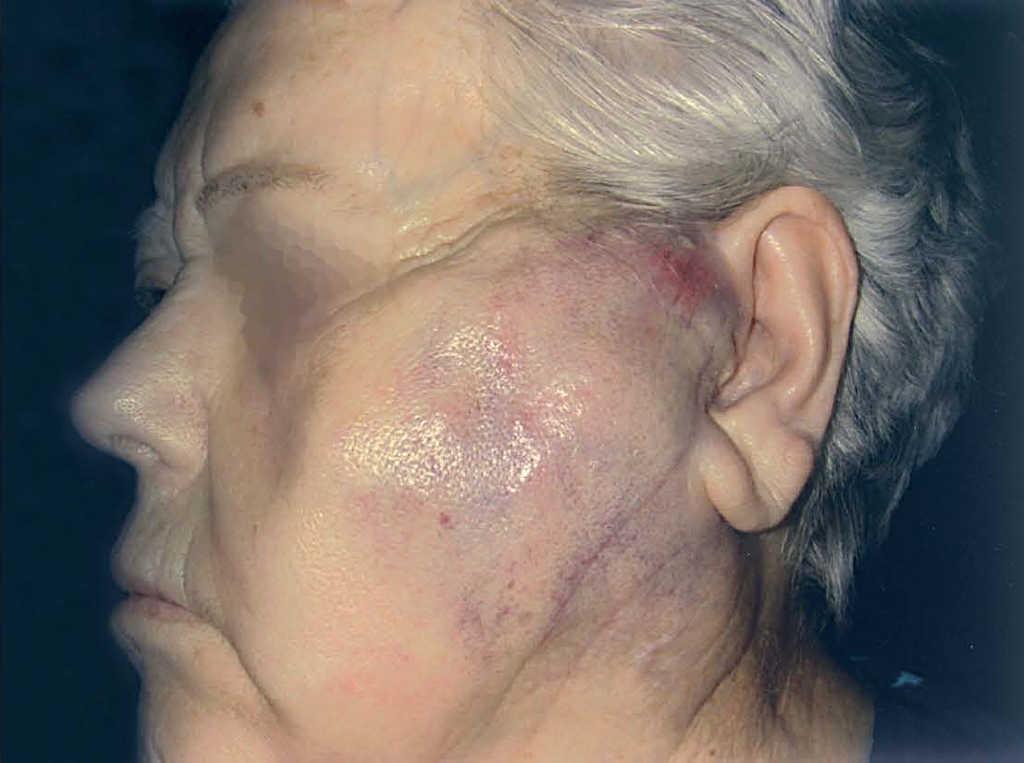
Fig. 2.—Placa violácea de bordes mal definidos.
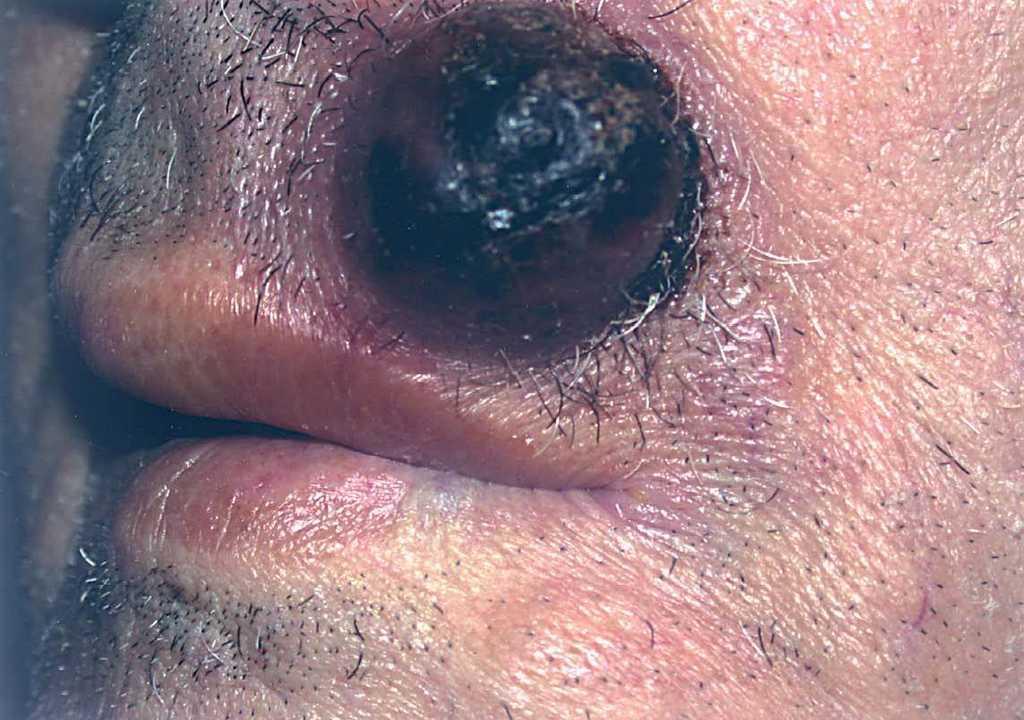
Fig. 3.—Nódulo violáceo con áreas necrosadas.
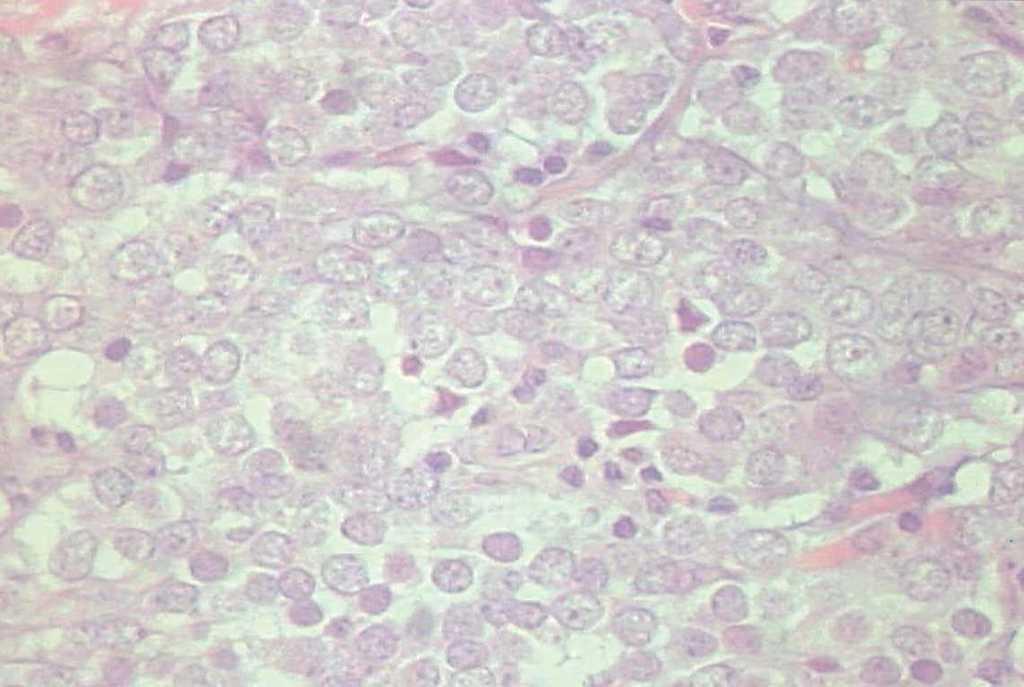
Fig. 4.—Células pequeñas de citoplasma inaparente y núcleo ovalado con cromatina dispersa. (Hematoxilina-eosina, ×200.)

Fig. 5.—Enolasa neuroespecífica. Positividad citoplasmática difusa. (Hematoxilina-eosina, ×200.)
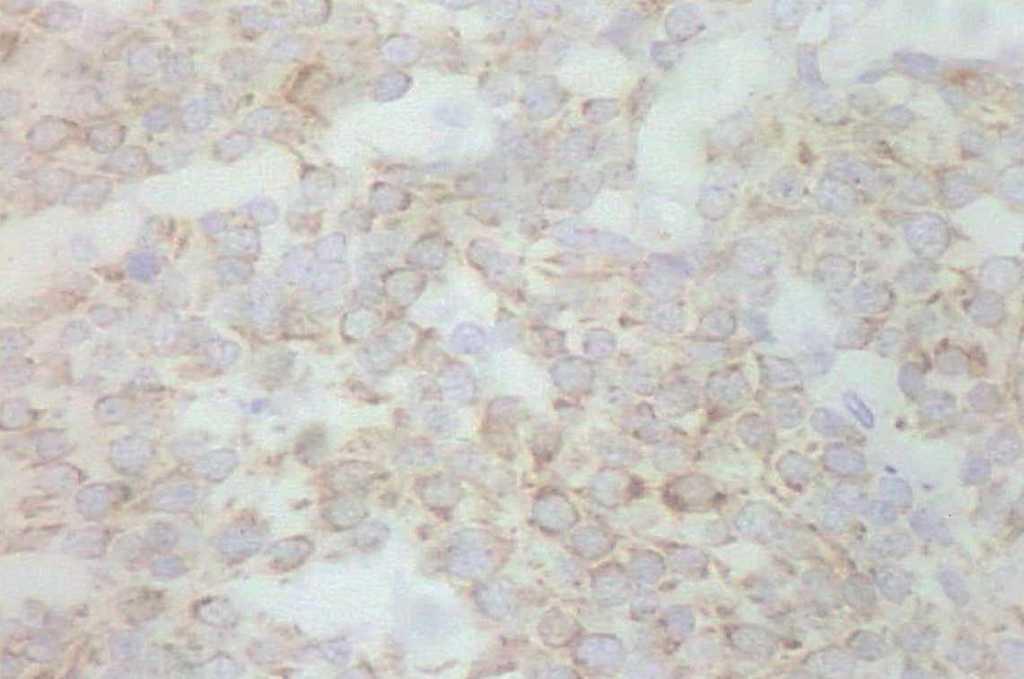
Fig. 6.—Citoqueratinas AE1/AE3. Motas perinucleares. (Hematoxili-na-eosina, ×200.)
Los 4 pacientes con lesiones nodulares fueron tratados con cirugía. Dos de ellos presentaron recidivas que fueron posteriormente extirpados. La otra paciente ingresó por disfagia secundaria a las adenopatías submaxilares. Cuando se le iba a realizar estudio de extensión, falleció debido a una neumonía por estafilococo dorado resistente a meticilina. Sólo un paciente recibió terapia adyuvante con quimioterapia (6 ciclos con carboplatino y etopósido durante 4 meses) y seguidamente radioterapia (50 G en 20 sesiones durante mes y medio en región pélvica).
DISCUSIÓN
El CCM tiene una incidencia en Estados Unidos de 0,23 por 100.0005 . Suele aparecer en la séptima u octava década de la vida en pacientes de raza blanca, sin haber un claro predominio de sexo6 . Se ve preferentemente en cabeza y cuello (43 %), un 20 % del total en región periorbitaria3,4 , aunque puede aparecer en extremidades (35 %) y con menos frecuencia en nalgas (14 %) y tronco (7 %)7 . Dado que aparece en zonas fotoexpuestas y que se asocia con otros procesos cutáneos relacionados con el daño actínico como queratosis actínicas, carcinoma espinocelular, carcinoma basocelular o enfermedad de Bowen, se piensa que en su etiología pudieran estar implicados los rayos ultra-violeta3,6 . También se ha descrito en inmunodeprimidos y recientemente se ha visto que puede estar asociado a otros tumores en un 25 % de los pacientes, como de mama, ovarios y cabeza y cuello8 .
La forma de presentación clínica se muestra como una tuberosidad indolora de 0,5 a 5 cm rosácea de aspecto brillante, con telangiectasias en superficie, que puede evolucionar a una placa violácea mal definida. El tumor evidencia un comportamiento rápido, ya que entre la mitad y dos terceras partes de los pacientes presentan adenopatías y altamente recidivante, con una tasa del 33 % en el primer año10 . La supervivencia a un año es del 88 %, a los 3 años, del 55 % y a los 5 años, del 30 %.
El diagnóstico definitivo viene determinado por la inmunohistoquímica. El tumor muestra afinidad por marcadores neuroendocrinos (cromogranina, sinaptofisina, enolasa neuroespecífica) y epiteliales (citoqueratinas 8, 18, 19 y 20). Estos últimos se disponen en forma de gruesas motas perinucleares típicas. En nuestros casos se utilizó un combinado de citoqueratinas de alto y bajo peso molecular (AE1/AE3). No se aprecia inmunotinción para antígeno leucocitario común, S-100, integrina o proteína gliofibrilar7 .
La histogénesis del CCM es desconocida. Entre las posibles células primarias se encuentran la célula de Merkel epidérmica, por sus similitudes ultraestructurales e inmunohistoquímicas, un equivalente dérmico de la propia célula de Merkel, dado que el tumor infiltra sólo la dermis, una célula del sistema APUD procedente de la cresta neural o una célula madre epidérmica o anexial. La fuerte positividad a determinadas citoqueratinas y la existencia, en ocasiones, de zonas tumorales con diferente diferenciación apoyan esta última hipótesis7 . El cambio citogenético más frecuente es la pérdida de heterozigosidad debida a traslocaciones y deleciones en el cromosoma 1. Se aprecian anomalías semejantes en tumores originados en la cresta neural como melanoma, neuroblastoma y feocromocitoma10 .
Como factores de mal pronóstico se consideran un tamaño del tumor de más de 2 cm, sexo masculino, edad mayor de 60 años, existencia de afectación vascular, linfática o metástasis, presencia de otro tumor no cutáneo8 y localización en tronco4,5 . También se postuló que los tumores en cabeza y cuello presentaban peor pronóstico, quizás a causa de los márgenes menos amplios realizados con el fin de evitar defectos cosméticos4 . Se ha sugerido que la expresión del CD4411 y una estroma rica en laminina, colágeno tipo IV e integrinas12 podrían correlacionarse con el riesgo de metástasis, mientras que la expresión del Bcl-2, p5313 y c-myc14 no se han relacionado con este hecho. Las neoplasias cutáneas asociadas no tienen mal pronóstico9 .
En lo referente al tratamiento, no hay protocolos establecidos debido a la escasa incidencia del CCM. En la tabla 2 se muestran tratamientos propuestos por algunos autores15 para los diferentes estadios. Nuestros pacientes fueron tratados con cirugía. Únicamente en el cuarto caso se recurrió a tratamiento adyuvante con quimioterapia y radioterapia. Respecto a la cirugía, existe controversia entre la cirugía de Mohs, ideal en procesos recidivantes16 , y la extirpación con margen amplio de 2-3 cm, la más empleada. También está en discusión, al igual que en el melanoma, la utilidad del ganglio centinela17 . La gammagrafía por octreótida no ha sido eficaz en la detección de metástasis18 . La radioterapia previene recidivas locales y actúa sobre las adenopatías locorregionales3,5,6,17 , por lo que se recomienda siempre irradiar la zona del tumor y las cadenas de ganglios linfáticos sobre las que drene 15 . Se suelen seguir ciclos de 40-50 Gy en 20-25 sesiones durante unas 5 semanas. Mortier et al 19 trataron 9 pacientes en estadio I sólo con radioterapia. Las dosis fueron 60 Gy en 30 sesiones, abarcando la radioterapia zona del tumor, ganglios locales y otros campos. Sorprendentemente, no se apreció ninguna recidiva, por lo que la radioterapia demostró ser una técnica bastante eficaz. El CCM es un tumor quimiosensible, aunque no se ha comprobado que los quimioterápicos mejoren la supervivencia de los pacientes 6 . Se utilizan los mismos fármacos que en el carcinoma de células de avena (oat-cell) del pulmón, es decir, etopósido, carboplatino, adriamicina y ciclofosfamida, entre otros 5 .

TABLA 2. ESTADIO Y TRATAMIENTO
Dado su alto grado de recidivas, cobra gran importancia el seguimiento del paciente. Como sucede con el tratamiento, no se dispone de protocolos reglados. Algunos autores aconsejan revisiones cada mes, los primeros 6 meses, cada 2 a 3 meses durante 2 años y posteriormente cada 6 meses. Se realizaría en cada visita exploración física, analítica y pruebas de extensión4 .
Correspondencia:
Josep Bertó. Servicio de Dermatología. Hospital Clínico San Carlos. Doctor Martín Lagos, s/n. 28040 Madrid. España. josepberto@yahoo.es
Recibido el 25 de agosto de 2004. Aceptado el 12 de noviembre de 2004.


