








INTRODUCCIÓN
Las calcificaciones cutáneas se producen por el depósito anormal de sales insolubles de calcio en la piel, las que pueden disponerse desorganizadamente (calcificación) o como formación de hueso (osificación). Se componen de una fase orgánica constituida por colágeno y tejido elástico y una fase sólida formada por cristales de hidroxiapatita y fosfato cálcico amorfo (1).
El calcio y su metabolismo
El calcio está presente en el organismo, principalmente en forma de fosfato de calcio en el hueso. Aunque los niveles de calcio en el líquido extracelular son mínimos, su concentración es crítica para varias funciones y permanece muy constante, siendo regulada principalmente por la hormona paratiroidea a través de una serie de procesos que movilizan el calcio desde y hacia el líquido extracelular. Esta hormona actúa directamente en el hueso y riñón e indirectamente en la síntesis de la 1,25 dihidroxivitamina D3 en el intestino para elevar los niveles de calcio sérico. A su vez, la producción de hormona paratiroidea está regulada por la concentración de calcio libre extracelular.
El fósforo es un componente fundamental del hueso y es el principal anión de los fluidos intracelulares, cumpliendo funciones muy importantes en diferentes procesos enzimáticos celulares. La regulación del metabolismo del calcio y del fósforo, así como el de la vitamina D, hueso y dientes, está mediada por las hormonas paratiroidea y calcitonina que actúan generalmente en forma antagónica en la homeostasis de dichos iones.
El calcio es un catión divalente importante en diversos procesos fisiológicos del organismo, como neurotransmisión, coagulación sanguínea y contracción muscular y miocárdica; participa además en el crecimiento y diferenciación epidérmicos y modula la interacción entre algunas moléculas de adhesión (2).
CALCIFICACIÓN CUTÁNEA
También conocida como calcinosis cutis, etiopatogénicamente se clasifica en tres grupos principales: calcificación distrófica (producida por daño tisular secundario a procesos traumáticos, inflamatorios, degenerativos, neoplásicos, etc.), calcificación metastásica (asociada a diversos trastornos que cursan con elevación de los niveles plasmáticos de calcio y/o fósforo) y calcificación idiopática (no relacionada con daño tisular ni trastornos metabólicos) (tabla I).

Calcificación distrófica
Es la más frecuente de las calcinosis cutis y cursa con niveles plasmáticos normales de calcio y fósforo. Las lesiones aparecen en zonas previamente dañadas, ya por procesos inflamatorios, traumáticos o neoplásicos, o asociadas a otras enfermedades que presentan daño tisular. Se cree que el proceso se desencadena por liberación de fosfatasas alcalinas y acúmulo de mucopolisacáridos ácidos o bien porque la necrosis celular crea un ambiente carente de inhibidores de la calcificación (2). A nivel celular la mitocondria posee gran afinidad por el calcio, pudiendo concentrarlo, permitiendo su cristalización.
Cuando los depósitos de calcio son pequeños y localizados, limitándose a la piel y tejido subcutáneo, se denomina calcinosis circunscrita. Si los depósitos son de mayor tamaño o diseminados, comprometiendo fascias, músculos y tendones, además de la piel, se denomina calcinosis universal.
Calcificación distrófica localizada
Puede verse en diversos trastornos de tipo inflamatorio como acné, cicatrices de quemaduras (3), queloides (4), flebitis (5) y úlceras de piernas (6). Algunos agentes infecciosos pueden producirla, particularmente algunas enfermedades parasitarias como la oncocercosis, cisticercosis, dracunculosis, loiasis, filariasis y quiste hidatídico (7); más raramente se la ha descrito como complicación de una infección intrauterina por virus herpes simple (8).
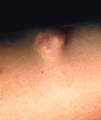
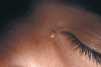
También puede ocurrir en asociación con múltiples neoplasias, benignas y malignas, apareciendo habitualmente en el tejido que rodea al tumor. Entre las primeras destaca el pilomatricoma (Fig. 1), que en el 75% de los casos aparece calcificado; en los pilomatricomas perforantes puede ocasionalmente verse la eliminación de material cálcico a través del tumor mediante un proceso de eliminación transepidérmica, posiblemente originado en la masa tumoral que actuaría como cuerpo extraño (9). Entre las malignas, en un estudio de 200 carcinomas basocelulares 41 casos (21%) mostraron evidencias de calcificación distrófica, en contraste con seis casos (3%) en carcinomas espinocelulares; los basocelulares calcificados mostraron un subtipo histológico más agresivo y tendían a localizarse en el tronco (10).
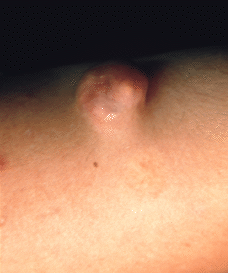
FIG. 1.--Pilomatricoma. Lesión de consistencia dura, lobulada y de coloración amarillenta.
Las calcinosis de origen traumático pueden verse tras diversos tipos de injuria tisular, tales como inyecciones (11), infiltraciones (12, 13), administración de soluciones intravenosas de calcio (14-16) con o sin extravasación de las mismas, punción con aguja en forma repetida (17, 18), radioterapia (19), registro de potenciales evocados (20), electromiografía (21) y electroencefalografía, que por ser la más estudiada revisaremos más en detalle.
Calcinosis cutis tras electroencefalografía: por sus cualidades hidroscópicas y por ser un buen electrólito el cloruro de calcio está incorporado en algunas pastas conductoras empleadas en electroencefalografía; además al ser un irritante primario de la piel contribuiría en producir una menor resistencia y un mayor contacto eléctrico (22). La técnica habitual para la realización del examen incluye provocar una ligera abrasión de la piel en los lugares de colocación de los electrodos, lo que permitiría un implante directo del cloruro de calcio en esas zonas (23), produciendo la calcinosis cutánea.
Las lesiones aparecen a los pocos días en forma de pápulas blanquecinas de superficie costrosa o verrugosa parecidas a quistes de milio (24), localizadas en el cuero cabelludo o lóbulos auriculares, exactamente en los sitios de colocación previa de los electrodos.
Al estudio histopatológico pueden observarse depósitos de calcio en la dermis superior, inmediatamente por debajo de la epidermis, junto a una reacción granulomatosa a cuerpo extraño. La epidermis puede aparecer engrosada o hiperqueratósica, con áreas de edema o degeneración en las que pueden detectarse partículas de calcio al igual que en algunos folículos pilosos (22).
Las lesiones pueden ser reproducidas experimentalmente traumatizando repetidas veces la piel con cinta adhesiva y luego aplicando en la zona pastas que contengan cloruro de calcio (23, 25). Su aparición puede prevenirse empleando pastas para electrodos que no contengan cloruro de calcio o evitando la abrasión previa de la piel (23).
Calcificación distrófica asociada a otras enfermedades
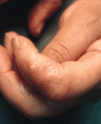
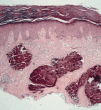
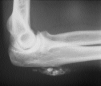
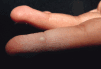
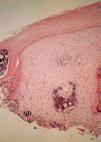
Algunas enfermedades del tejido conectivo pueden asociarse a calcificación distrófica, especialmente la esclerodermia y la dermatomiositis (26, 27). En la primera, la calcificación es habitualmente de pequeña cuantía y aparece usualmente en la piel y tejido celular subcutáneo de las manos; en el síndrome CREST la calcinosis es un rasgo fundamental para su diagnóstico e inicia el acrónimo, con lesiones de aparición insidiosa localizadas principalmente en las extremidades superiores (28) y en especial en los dedos de las manos (Fig. 2), las que pueden ulcerarse y dar salida a un material grumoso calcáreo. Los depósitos de calcio se presentan como nódulos de 1 mm a 2 cm de diámetro o como microdepósitos en la dermis normal en forma de microcristales perifibrilares o cristales en forma de aguja (29). Su aspecto radiológico es característico (Fig. 3), por lo que raramente se biopsian (30) (Fig. 4).

FIG. 2.--Síndrome CREST. Nódulos duros en los dedos que dan salida a material grumoso calcáreo.
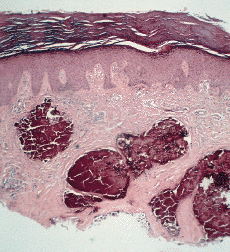
FIG. 3.--Aspecto radiológico del síndrome CREST.

FIG. 4.--Síndrome CREST. Depósitos de calcio en la dermis.
En la dermatomiositis la calcificación suele ser más extensa y se localiza principalmente en la musculatura del hombro o pelviana (31); se asocia más frecuentemente con la forma juvenil de la enfermedad (apareciendo hasta en el 60% de los casos) que con la de inicio en la edad adulta, en la que sólo ocurre en menos del 10% de los pacientes (32).
La calcificación en el lupus eritematoso sistémico es mucho más rara (33, 34) y habitualmente se circunscribe a la dermis o tejido subcutáneo afectados por la enfermedad, aunque también se la ha observado en tejidos blandos más profundos, libres de lesiones cutáneas. Muchas veces constituye un hallazgo radiológico con cierta predilección por las extremidades y las nalgas (35); en algunos casos la calcificación se ha limitado a las áreas que presentaban lesiones discoides y más raramente ha precedido en años al inicio de la enfermedad (36). En un caso su aparición se relacionó con el uso de hemodiálisis por una nefritis lúpica, con depósitos de tipo cistoide de inicio precoz (37).
Se presume que el origen de las calcificaciones en todos estos pacientes se producen por el daño tisular, en el que el fosfato unido a las células necróticas o a las proteínas desnaturalizadas serviría como base para el depósito de calcio (35).
Las enfermedades del colágeno pueden evolucionar al desarrollo de paniculitis, las que eventualmente también pueden mostrar calcificación distrófica.
La paniculitis lúpica o lupus profundo se caracteriza por nódulos subcutáneos localizados en la cabeza, porción proximal de las extremidades o en el tronco, los que pueden ulcerarse y mostrar calcificación; al curar y cicatrizar característicamente originan lipoatrofia (38).
Otra forma de paniculitis que puede calcificarse es la de origen pancreático, la que aparece en casos de pancreatitis o de adenocarcinoma pancreático; las lesiones aparecen en brotes de nódulos eritematosos dolorosos de localización pretibial, presumiblemente producidos por la liberación de enzimas pancreáticas que actúan sobre la grasa subcutánea.
La necrosis grasa subcutánea del recién nacido puede ocasionalmente calcificarse y en algunos casos evolucionar a hipercalcemia sintomática (39). Se manifiesta clínicamente con nódulos y placas eritematosas localizadas en las mejillas, espalda, nalgas y extremidades, que aparecen durante las primeras semanas de vida (40).
En el síndrome de Ehlers-Danlos, especialmente en el tipo 1, puede haber pequeñas esferas subcutáneas calcificadas; se cree que representan lóbulos de grasa isquémica calcificados y aparecen hasta en el 30% de los casos, generalmente sobre prominencias óseas de las extremidades (41).
En el síndrome de Werner, un trastorno hereditario autosómico recesivo que origina envejecimiento precoz, puede haber calcificación de tejidos blandos, especialmente vasos, ligamentos, tendones y celular subcutáneo; la enfermedad muestra una alta incidencia de neoplasias y muerte en edades tempranas de la vida (42).
El pseudoxantoma elástico es una enfermedad hereditaria rara en que aparecen calcificaciones de las fibras elásticas de la piel, arterias y membrana de Bruch ocular, por anomalías en las fibras de elastina; esto se traduce en diversas complicaciones que incluyen hipertensión arterial, hemorragias digestivas, oclusión coronaria, claudicación intermitente y prolapso de la mitral (43).
En la porfiria cutánea tardía puede ocurrir calcificación distrófica de las placas esclerodermoides. Las lesiones aparecen con mayor frecuencia en las regiones preauriculares, cuello y dorso de las manos (2). Pueden acompañarse de eliminación transepidérmica de sales de calcio (44).
Calcificación metastásica
Se produce en relación a procesos que cursan con niveles plasmáticos elevados de calcio y/o fósforo.
Infrecuentemente se han descrito casos de calcificación cutánea que cursan con niveles plasmáticos de calcio normales sin que se detecte enfermedad alguna asociada. Se presume que en estos casos, denominados idiopáticos, la calcinosis cutis puede ser un signo predecesor de una enfermedad subclínica subyacente, siendo muchos de ellos incluso cuestionables (45).
La calcificación de los tejidos subcutáneos es un proceso infrecuente y tardío en los pacientes nefrópatas crónicos; sin embargo, la calcificación de los tejidos blandos se ve en un 79% de los pacientes en diálisis y en un 44% de los pacientes no dializados (46), afectando principalmente el corazón, pulmón, estómago y riñones; en este mismo estudio, que incluyó a 56 pacientes dializados y 18 no dializados, sólo se detectó una calcificación cutánea (46). El proceso se considera tardío y los pacientes que lo presentan tienen una corta sobrevida, generalmente menos de 1 año, a menos que medie un tratamiento (47). La insuficiencia renal crónica lleva a un hiperparatiroidismo secundario que origina liberación de calcio de los huesos e inhibición de la reabsorción tubular de fosfato; el calcio sérico, sin embargo, no se eleva y el fosfato cálcico es depositado en distintos órganos. Cuando el producto de ambos (calcio * fósforo), que normalmente es de 40, está elevado del orden de 65 a 70, se produce el depósito subcutáneo de fosfato de calcio (48). Este cuadro clínico es denominado hiperparatiroidismo renal. Los depósitos subcutáneos se han descrito como pápulas o placas localizadas en la cara interna de los muslos, en la línea axilar anterior o posterior, fosa antecubital y poplítea, periné, escroto, así como también alrededor de las grandes articulaciones (49, 50).
En el hiperparatiroidismo primario, generalmente producido por un adenoma de las paratiroides, la hormona paratiroidea es secretada en exceso, originando una elevación del calcio plasmático. Sin embargo, la calcificación metastásica sólo se produce en etapas tardías, cuando el calcio precipita en los túbulos renales originando una insuficiencia renal y un aumento en el fosfato sérico, permitiendo la precipitación del fosfato cálcico en los tejidos blandos (47). Esta secuencia de eventos es probablemente la misma que se produce en la hipervitaminosis D, en el síndrome leche-álcalis, en la sarcoidosis y en otras enfermedades que cursan con destrucción ósea.
La ingesta crónica de dosis elevadas de vitamina D puede originar hipervitaminosis D. Sus síntomas y signos son debidos a hipercalcemia e incluyen debilidad, cefalea, náuseas, poliuria y nefrolitiasis (2). El componente local para que se produzcan los depósitos de calcio sigue siendo importante, ya que ocurre especialmente en zonas alteradas, como, por ejemplo, articulaciones deformadas por gota o artritis reumatoide (51) y no así en otras situaciones en que el producto del calcio y fósforo está elevado, pero no existen alteraciones anatómicas (52).
Hasta hace algunos años era muy común que los pacientes portadores de un síndrome ulceroso consumieran regularmente cantidades importantes de leche y álcalis, llegando algunos de ellos a presentar depósitos subcutáneos de calcio. La afectación cutánea no era constante; en algunos pacientes no había lesiones cutáneas, pero sí en otras zonas como el ojo (53); otros, sin embargo, presentaban grandes depósitos subcutáneos de calcio (54). Con la simple medida de eliminar la ingesta de álcalis y de leche se puede lograr una reducción gradual de las calcinosis (55).
Calcifilaxis
Seyle en 1962 describió una forma de calcificación de tejidos blandos de tipo experimental en la que los tejidos responden a una serie de estímulos formando depósitos de calcio (56, 57). Se la define como una calcificación localizada aguda que afecta a varios órganos, incluyendo la piel, riñones y menos frecuentemente el pulmón, corazón y tracto gastrointestinal.
Las lesiones cutáneas se caracterizan por áreas de coloración livedoide, sensibles, que se localizan en la región proximal de las extremidades. Evolucionan a placas induradas y nódulos bien delimitados que progresan a ulceraciones profundas con formación de escaras que en algunos casos han llevado a la amputación espontánea de dedos o extremidades (57).
El estudio histopatológico muestra depósitos de calcio en los vasos pequeños y medianos, acompañados de necrosis e isquemia epidérmicas. En un caso se ha descrito calcificación perineural (58).
Su etiopatogenia es incierta y probablemente sea multifactorial. La mayoría de los casos ocurren en pacientes con insuficiencia renal crónica e hiperparatiroidismo secundario (59), aunque se han descrito casos con función renal normal (60). Las alteraciones del metabolismo fosfocalcémico son inconstantes (58). Otros posibles orígenes son un aporte excesivo de vitamina D, hipertensión y alcalosis sistémica tras hemodiálisis. Los pacientes infectados por el virus de la inmunodeficiencia humana pueden evolucionar a insuficiencia renal y otras anomalías endocrinológicas, por lo que están predispuestos a padecerla (61). Más raramente se la ha descrito en relación a carcinoma de mama metastásico osteolítico (62).
El tratamiento sólo es de soporte, siendo importante la corrección de la hipercalcemia. Se ha utilizado la paratiroidectomía con buenos resultados (63). Se deben corregir las alteraciones hidroelectrolíticas y las infecciones. Si se detecta algún factor desencadenante (albúmina, corticoides, inmunosupresores, productos sanguíneos) se debe hacer lo posible por eliminarlo (57).
El pronóstico de estos pacientes es malo y la mayoría fallece de sepsis secundarias a infecciones de úlceras (64).
Calcificación idiopática
Se presenta sin una causa clara conocida y no se asocia a alteraciones en el metabolismo del calcio ni del fósforo. Existen cinco entidades que pueden incluirse en este grupo.
Nódulo calcificado subepidérmico
Fue descrito inicialmente por Duhring en 1877 con el nombre de cálculo cutáneo; en forma posterior Winer (65) lo individualiza como entidad y lo denomina calcificación nodular congénita solitaria de la piel al describir tres niños con lesiones únicas presentes desde el nacimiento. Observaciones subsecuentes que se presentaron con lesiones múltiples o con aparición en la edad adulta (66) llevaron a la preferencia de algunos autores por el término nódulo calcificado subepidérmico (67).
El origen del trastorno es desconocido. Se han postulado varias hipótesis en relación a su etiopatogenia, entre las que se incluyen la calcificación de un hamartoma de los ductos sudoríparos ecrinos (65), calcificación secundaria a necrosis grasa postraumática (68), calcificación de tecas de células névicas (69) o mesenquimatosas (66), depósitos cálcicos siguiendo a un proceso de degranulación mastocitaria de causa desconocida (70), asociación a niveles elevados de calcio en el sudor (67) y trauma en una deformidad congénita (71).
Clínicamente se manifiesta en forma de pápulas o nódulos cupuliformes de coloración blanquecinamarillenta y superficie lisa, verrugosa o mamelonada. Su consistencia es dura y en general no sobrepasan los 10 mm de diámetro. Habitualmente únicos, suelen localizarse en la cabeza --especialmente en las mejillas (72) (Fig. 5) u orejas (71-75) (Fig. 6)-- y menos frecuentemente en las extremidades (66) (Fig. 7); excepcionalmente se le ha descrito en la boca afectando gingiva o lengua (nódulo calcificado mucoso) (76) o en el pene (77). Por lo general hacen su aparición durante la primera infancia, aunque pueden ser congénitos o aparecer incluso en población geriátrica (78).
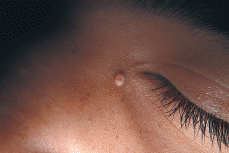
FIG. 5.--Nódulo calcificado subepidérmico.
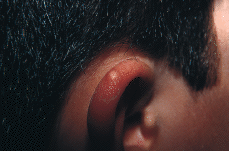
FIG. 6.--Nódulo calcificado subepidérmico.

FIG. 7.--Nódulo calcificado subepidérmico.
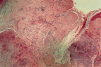
Histopatológicamente se observa material calcificado en la dermis superior en forma de masas homogéneas o agregados de glóbulos cálcicos rodeados por un infiltrado inflamatorio más bien escaso formado por macrófagos, células gigantes y mastocitos (Fig. 8). La epidermis suele ser hipertrófica y ocasionalmente puede mostrar fenómeno de eliminación transepidérmica (Fig. 9).
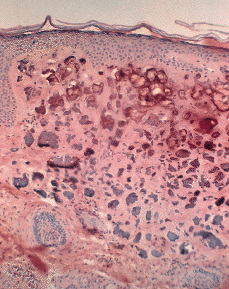
FIG. 8.--Depósitos de calcio en la dermis superior y reacción inflamatoria escasa en el nódulo calcificado subepidérmico.
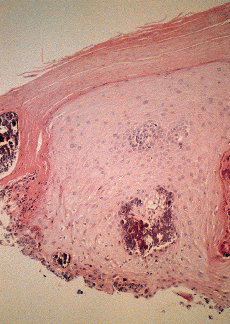
FIG. 9.--Fenómeno de eliminación transepidérmica en un nódulo calcificado subepidérmico.
El diagnóstico diferencial debe plantearse con molusco contagioso, verruga, cuerno cutáneo, xantoma (79) y con otras formas de calcinosis cutáneas.
El tratamiento consiste en su extirpación mediante curetaje bajo anestesia local, siendo raras las recidivas (72).
Calcificación de los cartílagos auriculares
Pese a que la calcificación de los cartílagos auriculares es considerado un fenómeno inusual, en un estudio se la detectó con una frecuencia de 3% mediante examen radiográfico de una serie de pacientes no seleccionados (80).
Clínicamente se produce un endurecimiento progresivo de los cartílagos auriculares sin afectación de la piel que los recubre. Suele ser asintomático, aunque existe imposibilidad para doblarlos o se les mueve como una unidad rígida que ha sido comparada a la bisagra de una puerta (81). Ocasionalmente se produce dolor a la compresión u otalgia en caso de afectación del conducto auditivo externo, la que se agrava por decúbito lateral (81). Los traumatismos y congelación son los factores etiopatogénicos más frecuentemente involucrados en su origen, aunque un número importante de casos --alrededor de un tercio-- son idiopáticos (80). Puede encontrársele asociada a otros múltiples procesos, entre los que se incluyen la insuficiencia suprarrenal, gota, hipopituitarismo, acromegalia, hipoparatiroidismo, hipertiroidismo, diabetes mellitus, prolactinoma, sarcoidosis, esclerodermia, periarteritis nodosa, síndrome de Tietze, osteodistrofia hereditaria de Albright, condromalacia sistémica, ocronosis, pericondrotis sifilítica, alteraciones en el metabolismo del calcio y del fósforo, radioterapia e hipersensibilidad al frío (81-84). Entre ellos, la insuficiencia suprarrenal es uno de los trastornos sistémicos más frecuentemente asociado a endurecimiento del cartílago auricular (85); su mecanismo etiopatogénico es desconocido, aunque se atribuye al propio déficit de cortisol (83).
Más raramente se ha descrito la presencia de focos de osificación de los cartílagos presumiblemente originados por exposición crónica al frío (86).
Calcinosis cutis de tipo miliar y síndrome de Down
Dentro de los diversos trastornos dermatológicos que han sido asociados al síndrome de Down, uno de reciente descripción es la calcinosis cutis idiopática miliar (87). Las lesiones aparecen en la infancia en forma de múltiples pápulas blanquecinas redondeadas, de 1 a 3 mm de diámetro, localizadas en la cara, dorso de las manos, dedos, palmas, plantas, muñecas, codos, muslos y rodillas (88). Algunos casos se han asociado a siringomas y a eliminación transepidérmica (88, 89) o más raramente a nevos múltiples del tejido conectivo (90).
El origen de las lesiones es desconocido y viene a sumarse a otras calcificaciones anormales que pueden observarse en el síndrome de Down, tales como las de los ganglios basales y de las válvulas cardíacas, lo que probablemente refleja un proceso de envejecimiento prematuro en esta enfermedad (87).
También se ha descrito una calcinosis cutis perforante en forma de granos de mijo en ingles y pubis de niños normales (91), sugiriendo alguna relación con la excreción de calcio en el sudor.
Calcinosis tumoral
Es una enfermedad hereditaria rara, de origen desconocido, en la que se desarrollan masas de gran tamaño generalmente sobre áreas de presión y articulaciones. Fue descrita por primera vez en 1898 por Duret, dándosele posteriormente diferentes nombres hasta que Inclan en 1943 la denominara calcinosis tumoral (92).
Es poco frecuente en Europa y Norteamérica; la mayoría de los casos descritos provienen de población negra de África, aunque en estos lugares también está subdiagnosticada, confundiéndose con otras patologías que presentan calcificación, tales como bursas calcificadas, tumores, tuberculosis, quistes, calcificación metastásica y oncocercosis (93).
Aparece generalmente en las primeras dos décadas de la vida, aunque se han descrito casos hasta los 79 años de edad, sin predilección por sexo (94, 95).
Clínicamente se caracteriza por la formación de masas calcificadas adyacentes a grandes articulaciones, pero sin llegar a comprometerlas. Son indoloras, únicas o múltiples, de crecimiento lento, pudiendo ser subcutáneas o intramusculares, y dependiendo de su localización estar firmemente adheridas a la fascia profunda. Son de consistencia firme, aunque en algunas áreas pueden ser de consistencia quística. Inicialmente son pequeñas, evolucionando posteriormente a grandes tumores lobulados. La piel que los recubre generalmente es normal, aunque puede ulcerarse, eliminando un material blanquecino e infectándose secundariamente (94-97). No hay compromiso de órganos internos y en la mayoría de los pacientes la calcemia es normal, a diferencia del fósforo que puede estar elevado.
Radiológicamente se caracteriza por la presencia de nódulos densamente calcificados en los tejidos blandos adyacentes a grandes articulaciones, tales como cadera, codo, escápula y sacro (93).
Al estudio histopatológico se distinguen dos etapas: inicialmente hay necrobiosis del colágeno que lleva a la formación de quistes asociada a una respuesta granulomatosa de tipo cuerpo extraño; posteriormente evoluciona a la etapa final de calcificación del colágeno, que primero es granular pero luego se forman depósitos densos que muy rara vez desarrollan metaplasia (93).
Su etiología es desconocida, planteándose dos hipótesis: una postula que se trataría de una forma de calcificación distrófica relacionada a injuria o trauma mecánico y la otra plantea que habría un trastorno metabólico del calcio y del fósforo (93).
El diagnóstico diferencial se plantea con calcinosis universal y circunscrita, intoxicación crónica por vitamina D, nefropatía crónica, síndrome de Barnett e hipeparatiroidismo primario (94, 97, 98).
El tratamiento consiste en la extirpación quirúrgica de las lesiones, aunque la recurrencia es frecuente, así como la aparición de nuevos focos de calcificación (94). La radioterapia no ha sido beneficiosa en la mayoría de los casos en que se ha empleado (99). Existe un caso descrito en que una dieta pobre en calcio y fósforo, asociada a hidróxido de aluminio, logró buenos resultados (100).
Calcificación escrotal idiopática
Trastorno infrecuente de origen desconocido caracterizado por depósitos de calcio a nivel de la dermis del escroto (101). Aparece en varones sanos durante la infancia o al inicio de la edad adulta, manifestándose clínicamente por pápulas o nódulos subcutáneos de localización escrotal que muestran una coloración blanquecinamarillenta (Fig. 10); las lesiones presentan una consistencia dura a la palpación y ocasionalmente eliminan un material blanquecino pastoso; generalmente son asintomáticas, aunque pueden acompañarse de una sensación de peso a nivel genital.
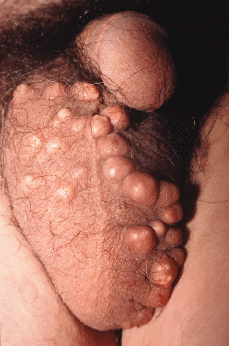
FIG. 10.--Calcificación escrotal idiopática. Múltiples nódulos de coloración blanquecinaamarillenta en la región escrotal.
La etiopatogenia del proceso es debatida, planteándose como posibles orígenes una calcificación distrófica del músculo dartros escrotal (102), calcificación distrófica secundaria a traumatismos mínimos en la zona (103), calcificación secundaria de tumores escrotales (esteatocistomas, quistes epidérmicos, fibromas, etc.), calcificación de nematodos muertos de Onchocerca volvulus en un caso (104) y calcificación distrófica de quistes miliares de los conductos ecrinos (105). La observación de casos que presentaban simultáneamente quistes epidermoides intactos, otros rotos y depósitos de calcio sin evidencias de pared epitelial, ha originado que la teoría más aceptada sea que la calcificación se origine en un quiste epidermoide que se inflama (106, 107); sin embargo, otros autores afirman que el trastorno es verdaderamente idiopático al detectar la presencia de queratina en la dermis adyacente a los depósitos de calcio mediante estudios inmunohistoquímicos (108). Al estudio histopatológico las lesiones se caracterizan por depósitos de calcio sin revestimiento epitelial y rodeados por una reacción inflamatoria de tipo cuerpo extraño (109).
El diagnóstico diferencial se plantea esencialmente con esteatocistomas y otros tumores de localización escrotal como xantomas, hidrocele o hematoma calcificado, etc.
El tratamiento es quirúrgico, extirpándose aquellas lesiones que produzcan mayores molestias.
OSIFICACIÓN CUTÁNEA
A diferencia de las calcinosis cutis en que los depósitos de calcio y fosfato se disponen en forma desorganizada dando salida a la extirpación a un material blando y pastoso que se desmorona, en la osificación cutánea la fase mineral se deposita en forma organizada, como si fuera hueso normal. Clínicamente las lesiones son duras y no se deshacen al aplastarlas, mostrando al estudio histopatológico una proliferación de tejido óseo con osteoblastos y a veces osteoclastos.
Las osificaciones cutáneas se dividen en primarias y secundarias (tabla II). Las formas primarias son muy infrecuentes y no se detecta en ellas alguna lesión cutánea previa; también se las denomina osteoma cutis, osteomatosis cutis u osteosis cutis. Las formas secundarias no son tan raras y se las puede encontrar en cicatrices quirúrgicas, enfermedades del colágeno y algunos tumores; en ellas la formación de hueso se produce por metaplasia de una lesión preexistente.

Osificación primaria
Osteodistrofia hereditaria de Albright
Descrita por Albright en 1952, se caracteriza por presentar múltiples áreas de formación ectópica de hueso que aparecen durante los primeros años de vida y que comprometen la piel, tejido subcutáneo, planos aponeuróticos, tendones y regiones periarticulares. El tamaño de las lesiones es variable, de unos pocos milímetros a 5 cm. En ocasiones pueden ulcerarse y dar salida a espículas óseas. La enfermedad se transmite con un patrón de herencia no bien establecido (110). Es más frecuente y severa en el sexo femenino.
Los casos afectados muestran una estatura baja, obesidad y cara redonda (111). Pueden haber anomalías craneofaciales que incluyen braquicefalia o más raramente microcefalia, frente prominente, puente nasal bajo, cuello corto e hiperostosis craneana. Los estudios radiográficos revelan depósitos óseos y calcificaciones de los ganglios basales y del plexo coroideo. Las anomalías dentales incluyen hipoplasia del esmalte y retardo en la dentición (112). La mitad de los casos muestran uñas anchas y cortas, especialmente a nivel del pulgar. El acortamiento de los metacarpianos, particularmente del cuarto y quinto dedos por cierre epifisiario prematuro, origina característicamente una ausencia de los dos últimos nudillos y su sustitución por hoyuelos a nivel de las respectivas articulaciones metacarpofalángicas, produciendo el llamado signo de Albright o signo del nudillo-nudillo-hoyuelo-hoyuelo. Una cuarta parte de los casos presentan cataratas o escleras azules y frecuentemente existe hipogonadismo o hipotiroidismo.
La enfermedad incluye dos variedades, el pseudohipoparatiroidismo, que se produce por una falta de respuesta a la hormona paratiroidea y se acompaña de hipocalcemia e hipofosfatemia, y el pseusoseudohipoparatiroidismo, que posee el mismo genotipo que el anterior, pero con niveles de calcio y fósforo normales (113). El 70% de los casos con hipocalcemia se asocia a retardo mental leve a moderado, el que es menos frecuente en los casos normocalcémicos.
Al estudio histopatológico pueden encontrarse espículas óseas de diverso tamaño en la dermis o en el tejido celular subcutáneo. Éstas contienen osteocitos, osteoblastos y muy raramente osteoclastos. A menudo pueden detectarse canales de Havers que contienen vasos sanguíneos y tejido conectivo; raramente se observan elementos hematopoyéticos.
El diagnóstico precoz de la enfermedad es importante para evitar los efectos derivados de la hipocalcemia. Con los años los pacientes tienden a hacerse normocalcémicos y la enfermedad tiende a aminorar.
Osteoma cutis
Se aplica este término a aquellos casos de osificación cutánea primaria en los que no se detectan evidencias de osteodistrofia hereditaria personal ni familiar. Se dividen en cuatro grupos:
-- Osteoma diseminado. Con lesiones múltiples presentes desde el nacimiento (114, 115).
-- Osteoma en placa. Con lesión única, grande, en forma de placa, presente desde el nacimiento y localizada en el cuero cabelludo, piel o tejido celular subcutáneo de una extremidad (116).
-- Osteoma solitario pequeño. Con lesión única adquirida, pequeña, con diversas localizaciones (Fig. 11), que en algunas ocasiones muestra eliminación transepidérmica de fragmentos óseos.
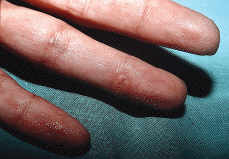
FIG. 11.--Osteoma solitario.
-- Osteoma miliar múltiple de la cara. Sólo aparece en mujeres jóvenes (117, 118) o mayores (119), mostrando una relación discutida con lesiones de acné vulgar (120). Clínicamente se presenta con lesiones papulosas pequeñas del color de la piel y duras al tacto, localizadas en la cara (121). En un caso se ha descrito exacerbación de las lesiones tras uso de isotretinoína para el tratamiento de un acné severo (122) y en otros pigmentación azulada de las mismas en relación a administración de tetraciclina (123) o minociclina (124).
Los rasgos histopatológicos en todas estas condiciones son similares a los de la osteodistrofia hereditaria (Fig. 12). El mecanismo etiopatogénico por el que se producen es desconocido, aunque se cree que cualquier injuria tisular que altere el pH, tensión de oxígeno, actividad enzimática y/o concentraciones de calcio y fosfato del medio, puede dar origen a calcificación y formación de hueso (125); sin embargo, en muchas ocasiones no puede detectarse ningún factor precipitante ni traumas. Se ha sugerido también que los osteoblastos se diferenciarían a partir de fibroblastos de la piel (126).

FIG. 12.--Osteoma solitario. Dermis con acúmulos de material osteoide con vaga disposición en trabéculas, algunas de ellas calcificadas; estroma mesenquimatoso fusocelular con diferenciación osteoblástica peritrabecular.
Heteroplasia ósea progresiva
Es un trastorno de la diferenciación mesenquimal que comienza en el período de lactancia, caracterizado por la osificación intramembranosa progresiva del tejido conectivo profundo, fascias, músculos y piel. Fue descrito por primera vez en 1994 (127).
Aparece casi exclusivamente en mujeres y es un proceso de osificación primaria, es decir, sin un fenómeno inflamatorio, tumoral ni traumático previo. La osificación empieza en la dermis para posteriormente comprometer tejidos más profundos, así como regiones adyacentes de la piel. Las lesiones se manifiestan precozmente, generalmente en el período neonatal, en forma asimétrica y con extensión progresiva. Clínicamente muestra placas constituidas por pequeñas pápulas que asemejan granos de arroz y que en su evolución aumentan de tamaño. Generalmente se distribuyen al azar en todo el cuerpo, aunque se han descrito casos unilaterales (128). No se asocia a otros rasgos dismórficos; sin embargo, la osificación progresiva lleva a deformidades ortopédicas importantes, como anquilosis de articulaciones y asimetría en la longitud de las extremidades.
Los exámenes de laboratorio son generalmente normales, aunque puede haber elevación de las fosfatasas alcalinas, dehidrogenasa láctica y creatinfosfoquinasa, que reflejan la osificación activa y el depósito de hueso en el músculo esquelético (129).
El estudio histopatológico muestra elementos maduros de hueso intramembranoso, cartílago, hueso encondral y depósitos de calcio en la piel, tejido conectivo profundo, fascias y músculos. La biopsia debe ser profunda.
La patogenia del proceso es desconocida. Se ha planteado que pueda deberse a que restos ectópicos de células mesenquimáticas normales se diferencien a osteoblastos (hamartomas) o a la estimulación de células mesenquimáticas normales extraesqueléticas a transformarse en osteoblastos (metaplasia). Muchos casos de osificación heterotópica pueden explicarse por alteraciones genéticas como el mosaicismo, apoyado en la distribución diseminada de las lesiones.
El diagnóstico diferencial incluye la osteodistrofia hereditaria de Albright, que rara vez provoca deformidades ortopédicas significativas y la fibrodisplasia osificante progresiva, que consiste en la osificación heterotópica de tejido conectivo profundo asociado a deformidades musculoesqueléticas; es un trastorno hereditario que comienza alrededor de los 5 años y progresa en forma simétrica y bilateral con mal pronóstico; la osificación no afecta la piel a menos que un algún espolón profundo 1a alcance (130); el acortamiento del ortejo mayor, presente en la gran mayoría de los casos, es un elemento importante en su diagnóstico, ya que está presente desde nacimiento.
El pronóstico es incierto y se relaciona con la localización y extensión de la osificación. No existe un tratamiento efectivo y la cirugía es la única alternativa en aquellos casos que presentan impotencia funcional, pudiendo llegar a requerir de la amputación de una extremidad en los casos más severos (127).
Osificación secundaria o metaplásica
Consiste en la formación de tejido óseo en una lesión preexistente, la mayoría de las veces de origen tumoral (131) o en cicatrices (132). El pilomatricoma es el tumor que más frecuentemente muestra osificación metaplásica, la que puede observarse en el 14-20% de los casos. A diferencia de la calcinosis que se inicia en el área de las células sombra, la osificación comienza en el estroma del tumor.
Tras excluir los pilomatricomas en una serie de 120 casos de osificación cutánea, cerca de la mitad de las lesiones restantes eran debidas a tumores (131). Otros tumores que pueden presentar osificación secundaria son el carcinoma basocelular (133) y menos frecuentemente el espinocelular (134), el siringoma condroide (135) y los nevos intradérmicos, en los cuales la osificación es a menudo secundaria a una foliculitis y son de localización facial (131).
La osificación secundaria de otros procesos inflamatorios, tales como esclerodermia (136) y dermatomiositis (137), es en general infrecuente. En 4 de 37 casos de condrodermitis nodularis helicis se detectó la presencia de focos de formación de hueso en la porción distal de la lámina condral (138); no existían antecedentes de trauma previo, sugiriéndose que el proceso inflamatorio crónico habría provocado la formación de hueso. Se han descrito osteomas faciales en un caso de foliculitis uleritematosa reticulada (139), no quedando claro si su aparición se relacionó con el empleo de dermabrasión. Recientemente se ha descrito un caso de osteoma cutis asociado a pseudoxantoma elástico (140).


