





INTRODUCCION
La acroqueratosis paraneoplásica (AP) es una dermatosis paraneoplásica poco frecuente caracterizada por la aparición de placas eritematosas y descamativas psoriasiformes en zonas acrales, principalmente manos, pies, nariz y pabellones auriculares. Todos los casos descritos en la literatura médica se asocian a malignidad, con una evolución paralela de la afectación cutánea y la neoplasia, por lo que se comporta como un verdadero marcador cutáneo de la enfermedad tumoral. Este hecho sugiere la existencia de algún factor patogénico común. Algunos autores defienden la existencia de un mecanismo inmunológico responsable de las lesiones cutáneas basándose en que la intensidad de las manifestaciones clínicas de la AP es proporcional a la concentración del antígeno del carcinoma de células escamosas (SCC-Ag) 1.
DESCRIPCION DEL CASO
Un varón de 64 años, con importante hábito tabáquico y enólico de largo tiempo de evolución consultó por una erupción pruriginosa eritematodescamativa que se había iniciado 2 meses antes en palmas y dorso de los dedos de las manos, y casi simultáneamente en las plantas y el dorso de los dedos de los pies.
La exploración dermatológica demostró la existencia de placas eritematosas con gruesa descamación y formación de fisuras en el dorso de los dedos de las manos (fig. 1). Las palmas y plantas presentaban un eritema difuso, y en el dorso de los pies se apreciaban lesiones ampollosas y costras serohemorrágicas adheridas secundarias a las ampollas (fig. 2). Todas las uñas de las manos y los pies eran distróficas, con onicólisis distal, hiperqueratosis subungueal y paroniquia. Ambos pabellones auriculares presentaban una fina descamación pitiriasiforme y, en el dorso de ambas rodillas, se observaban placas eritematoedematosas con pequeñas lesiones ampollosas en su superficie. La exploración física general objetivó la existencia de una notable ictericia cutaneomucosa y una hepatomegalia de tres traveses de dedos y de consistencia media.
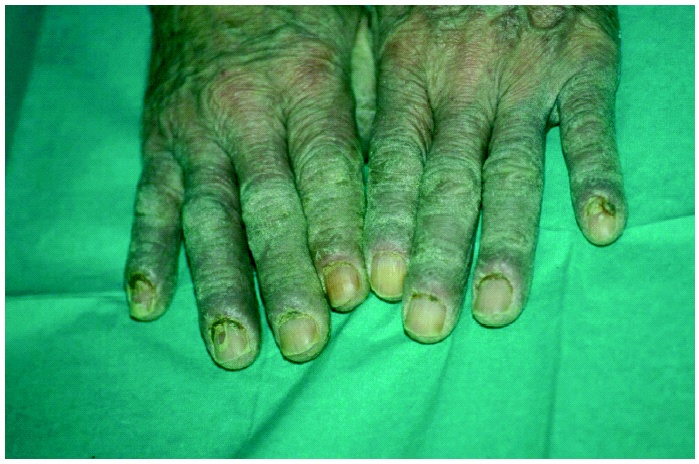
Fig. 1.--El dorso de los dedos presentaba placas eritematosas con gruesa descamación, engrosamiento de la lámina ungueal, onicólisis y coloración amarillenta de la misma.
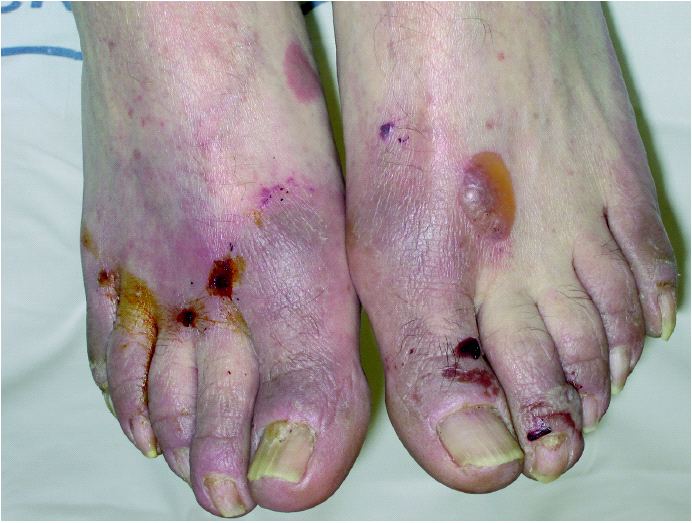
Fig. 2.--Placas eritematoedematosas con lesiones ampollosas y costras serohemorrágicas. Distrofia ungueal.
Los estudios analíticos mostraron una elevación de los marcadores de citólisis hepática y colestasis. La tomografía computarizada (TC) reveló, en la zona torácica, la existencia de una masa en la parte posterior de una costilla con derrame pleural bilateral asociado, sugestiva de lesión metastásica, así como un hígado graso en la zona abdominal. La endoscopia digestiva alta demostró la existencia de una masa tumoral en el tercio proximal del esófago, cuya biopsia fue informada como carcinoma epidermoide pobremente diferenciado.
Se realizaron dos biopsias cutáneas; una de dorso de dedo de la mano (fig. 3), que mostró una prominente hiperqueratosis y acantosis, con focos de paraqueratosis e hipogranulosis, algunas áreas de exocitosis y espongiosis, queratinocitos necróticos e infiltrado linfocítico perivascular. La segunda fue tomada de una lesión ampollosa localizada en la superficie de una placa eritematoedematosa del dorso de la rodilla. Su estudio histopatológico (fig. 4) mostraba un despegamiento subepidérmico con un infiltrado inflamatorio superficial de linfocitos, neutrófilos y eosinófilos, así como dermatitis de interfase con degeneración vacuolar de la capa basal. La inmunofluorescencia directa (IFD) de la piel perilesional de la segunda biopsia demostró la existencia de depósitos granulares de inmunoglobulinas (IgM, IgA) y C3 en la membrana basal (fig. 5).
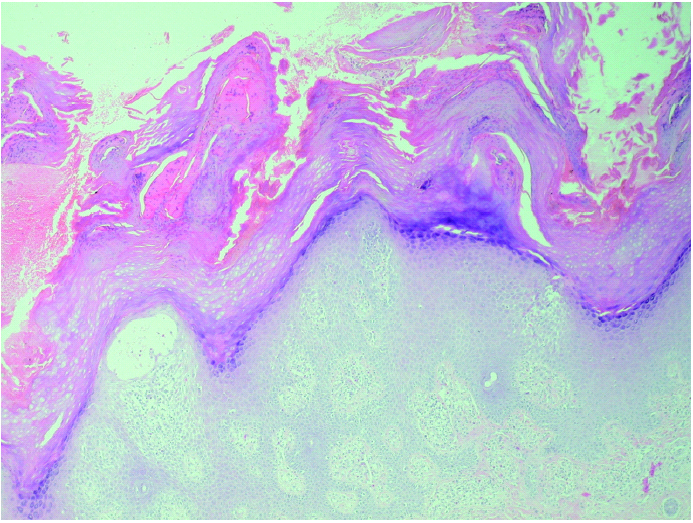
Fig. 3.--Hiperqueratosis paraqueratósica con acantosis, focos espongióticos e infiltrado inflamatorio perivascular. (Hematoxilina-eosina, ×40.)
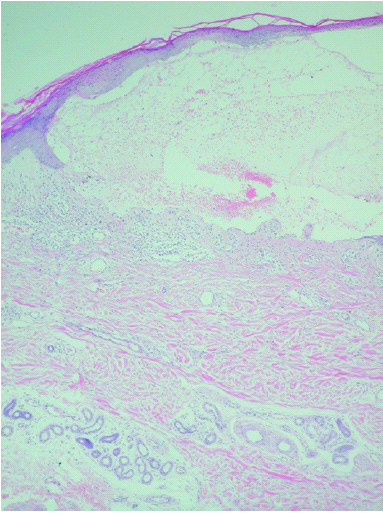
Fig. 4.--Ampolla subepidérmica con infiltrado inflamatorio superficial de linfocitos, eosinófilos y neutrófilos. (Hematoxilina-eosina, ×40.)

Fig. 5.--Depósito granular de C3 en membrana basal (piel próxima a una lesión ampollosa). (Inmunohistoquímica C3, ×400.)
Las lesiones descamativas desaparecieron parcialmente con tratamiento tópico con ácido salicílico al 10 % en vaselina. La evolución clínica después del diagnóstico fue desfavorable. El paciente fue trasladado al Servicio de Oncología, donde desarrolló episodios de hemorragia digestiva alta, con aparición secundaria de encefalopatía hepática; falleció 10 días después del traslado.
DISCUSION
Desde la primera descripción de la AP, se han descrito numerosos casos de esta dermatosis en la literatura médica, en su mayoría en relación con neoplasias malignas localizadas en la cavidad oral, faringe o laringe, esófago o pulmones, o incluso con adenopatías cervicales metastásicas de origen desconocido 2. No obstante, muchos otros tumores se han relacionado/asociado con este síndrome, como los de colon 3,4, de próstata 5, genitourinarios 6, de mama 7 e incluso la enfermedad de Hodgkin 8.
Clásicamente se han considerado tres fases en esta enfermedad 9. La primera se caracteriza por la aparición de eritema y descamación psoriasiforme en los dedos de las manos y pies, que afecta también a los pabellones auriculares y al dorso nasal, con alteraciones distróficas ungueales frecuentes (hiperqueratosis subungueal y onicólisis). En este momento la neoplasia suele ser asintomática. En una segunda fase, la erupción se extiende a palmas y plantas, mostrando una queratodermia difusa de tonalidad violácea, y coincidiendo frecuentemente con los primeros signos y síntomas de la enfermedad neoplásica. Finalmente, en un tercer estadio, la afectación cutánea se extiende a áreas proximales como rodillas, muslos y brazos, donde se muestran placas eritematosas mal definidas. En esta fase es frecuente constatar ya la existencia de metástasis ganglionares o viscerales.
Otras características clínicas menos frecuentes asociadas a este síndrome son ictiosis adquirida, hipopigmentación y vesículas o ampollas 3,10-13. En nuestro caso, se objetivaron lesiones ampollosas tensas en la superficie de las placas eritematoedematosas localizadas en el dorso de los pies y las rodillas, con un patrón clínico similar al de enfermedades ampollosas como el penfigoide ampolloso o la epidermólisis ampollosa adquirida. Sin embargo, los depósitos de membrana basal de IgA, IgG y C3 detectados mediante IFD mostraron un patrón granular no acorde con ninguna de ellas.
Bolognia et al 10, en una revisión bibliográfica de 93 casos de AP descritos en la literatura médica hasta 1995, encontraron lesiones ampollosas en 15 (17 %) casos. La biopsia de dichas lesiones en 6 pacientes mostró la existencia de ampollas subepidérmicas en cinco de ellos. Los estudios de IFD de 13 biopsias en 9 pacientes con AP descubrieron depósitos lineales de IgG, IgM, IgA y/o C3 en 2 de los 5 pacientes que presentaban ampollas además de las lesiones clásicas de AP. En los 7 pacientes restantes, la IFD tanto de las lesiones ampollosas como de las placas eritematodescamativas resultó negativa o mostró únicamente hallazgos inespecíficos. Pecora et al 11 describieron la presencia de un patrón difuso de depósitos de inmunoglobulinas (IgA, IgM y particularmente IgG) en biopsias de piel lesional (placas eritematodescamativas) y no lesional de pacientes con AP. Mutasim et al 12 hallaron depósitos inespecíficos de IgA y C3 a lo largo de la membrana basal en una biopsia de piel sana adyacente a una ampolla en un paciente con AP y lesiones ampollosas asociadas.
En la mayoría de los casos descritos de AP con lesiones ampollosas, éstas son subepidérmicas, aunque también se han documentado vesículas intraepidérmicas en algún caso aislado 13, probablemente secundarias al importante infiltrado inflamatorio linfocitario y edema en la dermis, así como a los fenómenos de exocitosis y espongiosis, que pueden aparecer en la AP. La IFD resultó negativa en este caso.
Aunque el mecanismo exacto de la formación de ampollas subepidérmicas en el contexto de la AP es desconocido, se han sugerido diversas hipótesis 11, como un cribado dermoepidérmico por un mecanismo similar al liquen plano (apoyado en la existencia de degeneración vacuolar de la capa basal en algunos casos), o la coexistencia de dos entidades distintas, por ejemplo AP y porfiria hepatocutánea tarda o penfigoide ampolloso o epidermólisis ampollosa adquirida (apoyado en la presencia de depósitos lineales en la membrana basal de IgG, IgM y/o C3 en algunos casos). Pecora et al 11 sugirieron la existencia de una respuesta inmunológica de tipo humoral a un antígeno neoplásico que presentaría reactividad cruzada con algún antígeno de membrana basal de la piel, que pondría en marcha una respuesta inmunológica, probablemente mediada por complemento, lo cual explicaría muchas de las características clínicas e histopatológicas descritas.
En nuestro caso, la presencia de alteraciones histopatológicas como focos necróticos en la epidermis, degeneración vacuolar de la capa basal, incontinencia pigmentaria y depósitos de inmunoglobulinas y C3 en la membrana basal, sugiere también la posibilidad de ataque inmunológico de la capa basal, probablemente como expresión de la existencia de reactividad cruzada entre antígenos del tumor y de la piel.
En definitiva, y aunque las lesiones ampollosas no se describen habitualmente como un hecho clínico típico de la AP, en nuestra opinión deben ser consideradas como parte del espectro clínico pleomórfico de esta dermatosis paraneoplásica, y muchas de las características histopatológicas asociadas apoyan una base inmunológica de esta entidad.
Correspondencia:
Miguel Cabanillas.
Departamento de Dermatología. Facultad de Medicina.
San Francisco, s/n.
15782 Santiago de Compostela. España.
mejaime@usc.es
Recibido el 10 de noviembre de 2005.
Aceptado el 13 de febrero de 2006.


