




Acral peeling skin syndrome (APSS) is a condition with autosomal dominant inheritance, caused by changes in gene TG5, responsible for the production of transglutaminase 5 (TG5).1 It presents at birth or in early childhood with the formation of blisters and subsequent peeling on the palms and soles. As the disease has a low incidence and little or no clinical repercussion, it tends to be underdiagnosed or incorrectly diagnosed as epidermolysis bullosa simplex, with which it shares certain characteristics.2
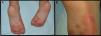
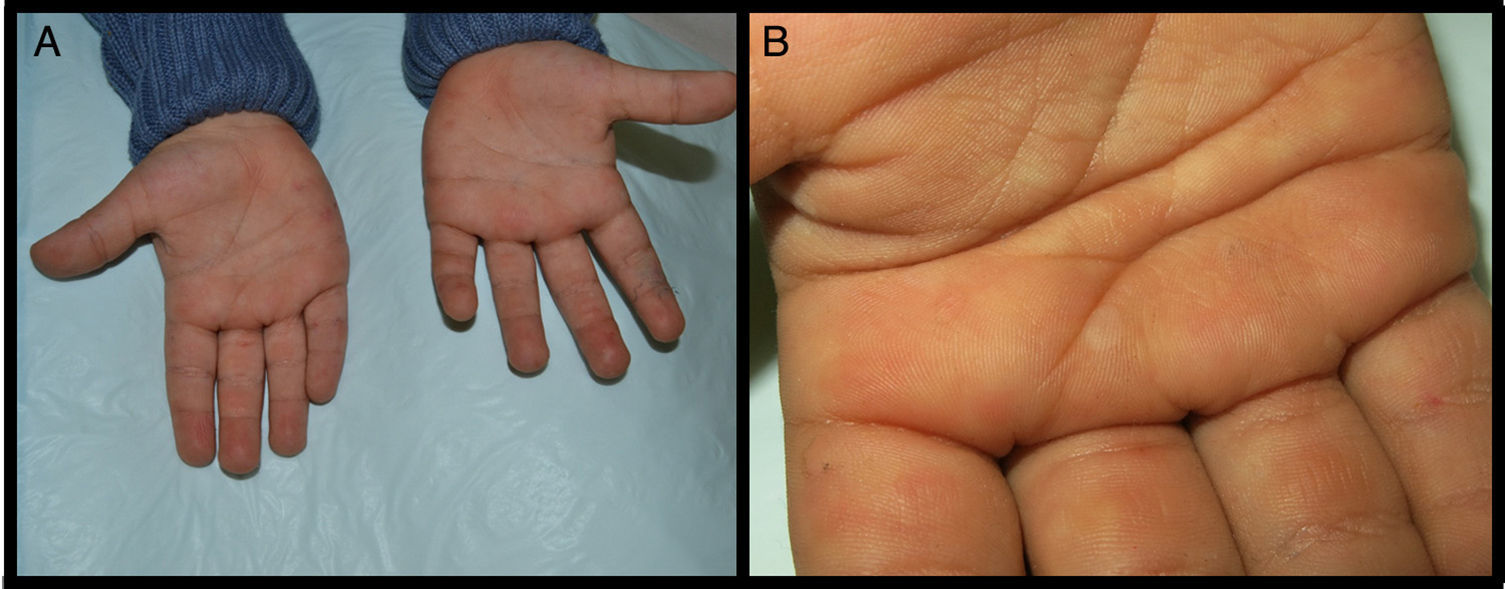
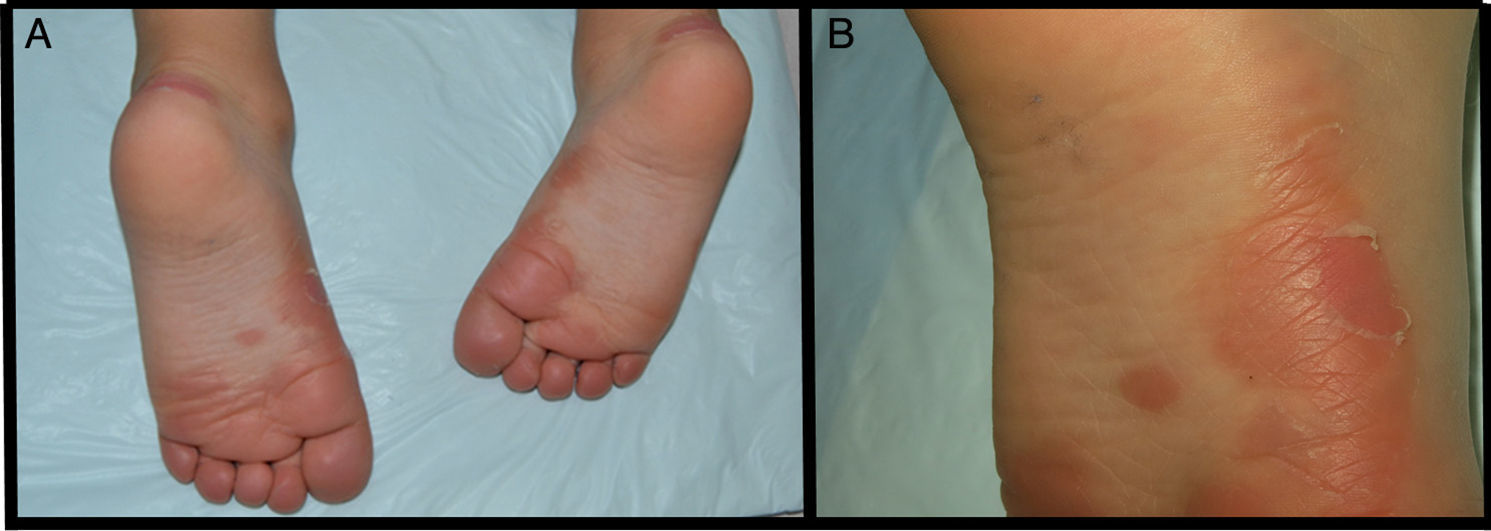
We present the case of a 3-year-old boy who, since 6 months of age, presented blisters with subsequent peeling on the palms and soles and, to a lesser degree, on the dorsum of the hands and feet. The episodes were asymptomatic and occurred with a frequency of once or twice a month; humidity exacerbated the condition. On physical examination, the patient presented tense and flaccid blisters in pressure areas, associated with peeling on the palms and soles (Figs. 1 and 2) and resolving areas with residual erythema. The rest of the skin and the mucosas were normal. Skin biopsy showed slight separation between the granular and corneal layers, with no inflammation (Fig. 3). In the stratum corneum, the intermediate cell layers above the plane of cleavage presented an unstructured appearance, with large round cells. The clinical and histopathologic findings were consistent with a diagnosis of APSS. Genetic analysis confirmed this hypothesis, as the patient presented 2 homozygous mutations of gene TG5: 1 pathogenic (P.G113C) and the other a polymorphism (P.T109H) frequently associated with the former.
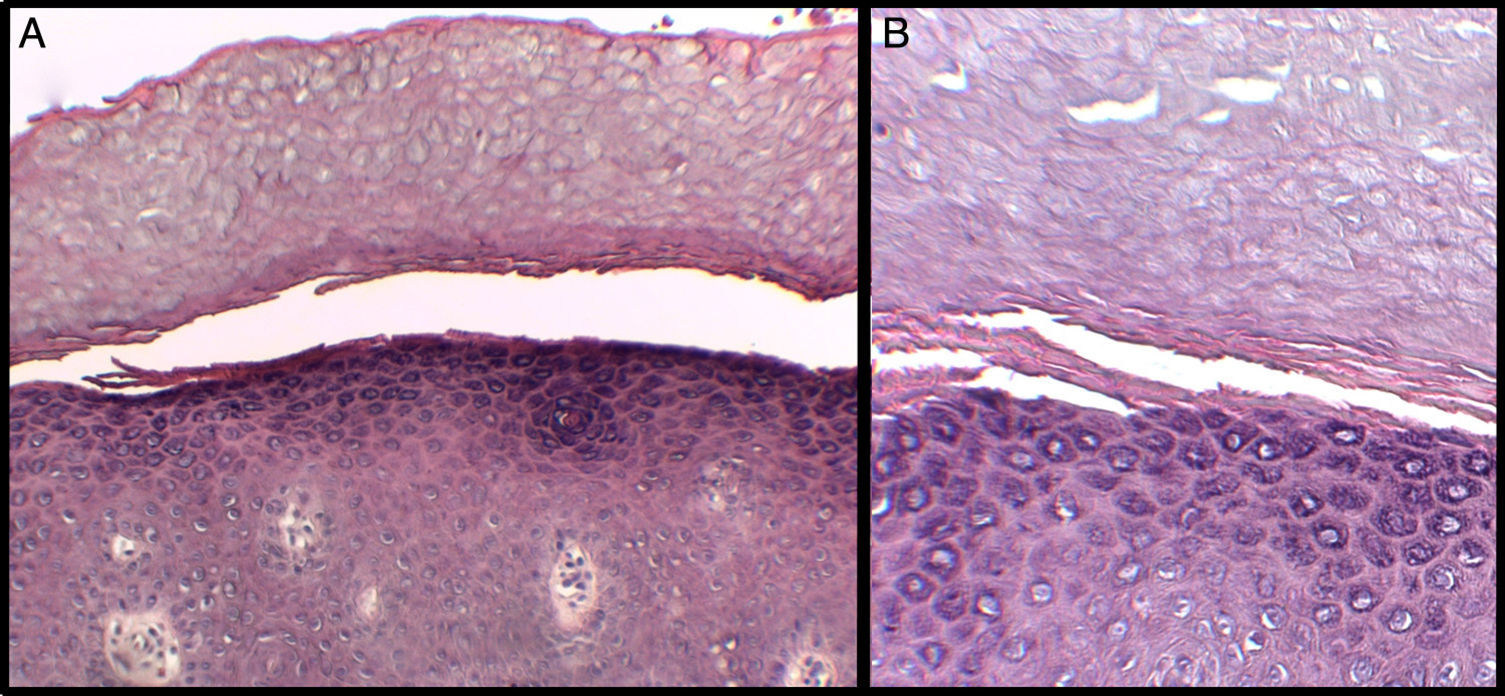
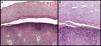
A, Separation between the corneal and granular layers of the epidermis. Hematoxylin and eosin (H&E), original magnification×10. B, Detail at higher magnification. Observe the appearance of the corneal layer above the plane of separation, with large round cells. H&E, original magnification×20.
APSS belongs to the group of peeling skin syndromes (PSS), which include a localized acral form (APSS) and a diffuse form.3–5 APSS was first described as a specific entity in 1982 by Levy and Goldsmith.6 Since that time, several individual case reports and case series of this syndrome have been published (Table 1). It characterized by the formation of blisters and superficial peeling of the skin of the palms and soles, leaving a painless residual erythema that heals without scarring. The manifestations are exacerbated by humidity and by physical factors such as trauma or friction. In general, symptoms are absent or mild. In addition, APSS presents a number of histopathologic characteristics. The main finding is separation between the granular and corneal layers. Another common feature, observed in the stratum corneum above the plane of separation with the granular layer, is the presence of several layers of cells of atypical appearance: large, round, pale cells with an edematous and less compact appearance. Electron microscopy provides a better image of the separation between the corneocytes in these layers, with fragmentation of the keratin filaments.7,8
Summary of the Most Relevant Findings of the Cases of APSS Published to Date.
| Countries/regions reported | Germany, United Kingdom, The Netherlands, Poland, Italy, Spain, Russia, Hungary, Slovakia, Finland, Sweden, East Africa, Tunisia, Middle East, Morocco, and Jordan |
| Age at onset of symptoms | From birth to 11 years |
| Age at time of diagnosis | From 1 to 47 years |
| Most common signs | Blisters and peeling of palms and soles, and dorsum of hands and feet |
| Exacerbating factors | Humidity, sweating, heat, friction, and trauma |
| Mutations most frequently reported | p.[Gly113Cys];[Gly113Cys] Heterozygous p.[Gly113Cys] p.[Met1Thr];[Met1Thr] p.[Lys445Asn];[Lys445Asn] p.[Val273Met];[Val273Met] p.[Thr109His];[Thr109His] |
Abbreviation: APSS, acral peeling skin syndrome.
The underlying genetic mechanism was elucidated by Cassidy et al.1 in 2005. Those authors identified the mutation responsible for the syndrome, localized in gene TG5 on chromosome 15q15. This gene codes a protein, TG5, whose function is altered in the patients with this syndrome.
TG5 is expressed in the corneal layer and is responsible for the formation of crosslinks between key proteins in the cornification process (loricrin, involucrin, filaggrin, and others); it is also essential for maintenance of an intact corneal layer.1,4,8 Analysis of healthy skin from individuals with APSS shows that the mutation is not exclusive to the acral skin areas, but it is hypothesized that the other transglutaminases present in the skin, in different proportions depending on the site, are sufficient to complete the cornification process. The absence of TG5 action cannot be sufficiently compensated in the skin of the palms and soles, where the corneal layer is thicker and is subject to greater pressures.9
Two mutations have been described that, when homozygous, give rise to the syndrome. The first of these is mutation G113C, which causes complete abolition of TG5 function. The other is T109M, which alone is not pathogenic, but is a polymorphism affecting the same allele, and that is frequently associated with the G113C mutation. This combination has been described in various patients from northern and central Europe, and it has been postulated that it is the result of a founder phenomenon in the European population. Fifty-nine cases with 15 new mutations affecting TG5 have recently been described,10 as well as a mutation of the protein cystatin A, alterations of which also cause manifestations of APSS.
Treatment is symptomatic, with emollients and measures aimed at reducing maceration and trauma.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Ours thanks to Javier Cañueto Álvarez of the Dermatology Department of Universidad de Salamanca, to Rogelio González Sarmiento and Elena Bueno Martínez of the Department of Molecular Medicine of Universidad de Salamanca, and to Luis Carlos Sáez Martín of the Dermatology Department of Hospital General Universitario Gregorio Marañón, in Madrid, Spain.
Please cite this article as: Ruiz Rivero J, Campos Dominguez M, Parra Blanco V, Suárez Fernández R. Síndrome de descamación de la piel acral: presentación de un caso y revisión bibliográfica. Actas Dermosifiliogr. 2016;107:702–704.


