






La enfermedad autoinflamatoria de la queratización (AiKD, por sus siglas en inglés) es un concepto clínico novedoso que engloba a las enfermedades que presentan antecedentes genéticos, así como mecanismos patogénicos mixtos de autoinflamación y autoinmunidad, lo que en su conjunto se traducirá en una queratinización aberrante de la piel. Los recientes avances han revelado causas genéticas y/o factores predisponentes para una serie de AiKD, dentro de los que se pueden enumerar la presencia de las mutaciones en el IL36RN, en relación con la psoriasis pustulosa, la acrodermatitis continua y la hidradenitis supurativa; en el CARD14, en relación con la pitiriasis rubra pilaris tipo V y algunas formas de psoriasis pustulosa, y en el NLRP1 en relación con la queratosis liquenoide crónica familiar (KLC, por sus siglas en inglés). Se sospecha que la fisiopatología de la AiKD también estaría presente en algunos trastornos no monogénicos. Se debe de comprender que existe una relación bidireccional entre la inflamación y la queratinización para poder determinar un tratamiento óptimo; así mismo para poder desarrollar nuevos fármacos ambos factores deben de tenerse en cuenta. Probablemente en los próximos años nuevas enfermedades inflamatorias de la queratinización serán incluidas dentro del grupo de las AiKD.
Autoinflammatory keratinization disease (AiKD) is a novel clinical concept encompassing diseases with a genetic background and mixed pathogenic mechanisms of autoinflammation and autoimmunity, leading to an aberrant keratinization of the skin. Recent advances in medical genetics have revealed genetic causes and/or predisposing factors for a number of AiKD's, such as mutations in IL36RN related with pustular psoriasis, acrodermatitis continua and hidradenitis suppurativa, in CARD14 in pityriasis rubra pilaris type V and some forms of pustular psoriasis, and in NLRP1 related with familial keratosis lichenoides chronica (KLC). It is suspected that AiKD pathophysiology would also be involved in non-monogenic disorders. The bidirectional relationship between inflammation and keratinization should be understood in order to outline optimal management, and new drug development should take both targets into account. We assume that new inflammatory keratinization diseases may be recognized as AiKDs in the coming years.
La enfermedad autoinflamatoria de la queratinización (AiKD, por sus siglas en inglés) es un concepto clínico novedoso que incluye enfermedades con antecedentes genéticos y mecanismos patogénicos mixtos de autoinflamación y autoinmunidad, que afectan a la epidermis y a la dermis superior, y que conducen a una queratinización aberrante de la piel. Las AiKD comparten una característica clínica principal: son lesiones cutáneas inflamatorias hiperqueratósicas, las cuales no responden de manera adecuada a las terapias convencionales. Las características clínicas son variables. Cuando la alteración de la queratinización afecta al epitelio folicular, las lesiones consisten en taponamiento y rotura folicular. La correcta definición de las AiKD puede ayudar al desarrollo de nuevas líneas de investigación, sobre todo con respecto a terapias dirigidas1.
En las últimas décadas, la comprensión de la inmunidad innata y adaptativa aplicada a las enfermedades dermatológicas ha demostrado importantes avances (fig. 1). No fue hasta 1999, como resultado del hallazgo en mutaciones en la línea germinal en la superfamilia 1 del receptor del factor de necrosis tumoral (TNFRSF1), responsable del síndrome periódico asociado al receptor del factor de necrosis tumoral (TRAPS), que se introdujo el concepto de autoinflamación. El término «autoinflamación» engloba un grupo de trastornos provocados por una disregulación del sistema inmunitario innato, y estas se deben de diferenciar de los síndromes autoinmunes2.

Algunas AiKD se consideraron inicialmente trastornos de la queratinización, como la alteración del metabolismo del ácido araquidónico en la psoriasis, lo que incentivó el tratamiento de esta entidad con retinoides orales. Como se observó que existía una participación de las células T, en los siguientes años estas se consideraron como trastornos autoinmunes y se desarrollaron las primeras terapias biológicas, destinadas a bloquear la activación de las células T o de las citocinas producidas por las células T. Recientemente, se ha postulado que la autoinflamación tomaría el papel principal en el inicio de estas enfermedades. El último concepto desarrollado de la AiKD agregaría nuevos mecanismos que deberían impulsar el desarrollo de nuevas terapias dirigidas.
La inmunidad innata se basa en el sistema de defensa, que clásicamente se ha descrito como un sistema precoz en la evolución, por lo cuál tiene una acción más rápida e inespecífica, así como no deja memoria inmunológica. Este sistema está compuesto por varios componentes: barreras físicas, receptores tipo toll, fagocitos, células asesinas naturales y mastocitos, siendo el complemento y los péptidos antimicrobianos los más importantes. La inmunidad innata y adaptativa actúan de manera coordinada, y la mayoría de los trastornos de la inmunidad son probablemente una combinación de disfunciones en ambos sistemas.
Enfermedades autoinflamatorias con afectación cutáneaLas enfermedades inflamatorias (AiD, por sus siglas en inglés) han despertado un interés creciente en la última década. Desde su definición inicial, se han identificado más de 30 genes nuevos asociados a enfermedades autoinflamatorias que afectan a diferentes partes del sistema inmunológico innato. Los primeros en incluirse fueron los síndromes hereditarios de fiebre recurrente como la fiebre mediterránea familiar (FMF) y el síndrome periódico asociado al receptor de TNF (TRAPS). Poco tiempo después, se agregaron otros síndromes febriles recurrentes, incluida la hiperinmunoglobulinemia D hereditaria con síndrome de fiebre periódica (HIDS) y el síndrome periódico asociado a criopirina (CAPS). Posteriormente, el concepto de autoinflamación se ha extendido a diversas enfermedades mendelianas, como el síndrome de Blau, síndrome de Majeed, deficiencia de antagonista del receptor de interleucina-1 (DIRA) y artritis piógena, pioderma gangrenoso y acné (PAPA), así como trastornos de etiología genética incierta que incluyen la fiebre periódica, la estomatitis aftosa, el síndrome de faringitis y adenitis (PFAPA), la enfermedad de Behçet, la enfermedad de Still, la enfermedad de Crohn y los síndromes autoinflamatorios adquiridos como el síndrome de Schnitzler2,3. Desde el punto de vista clínico, los síndromes autoinflamatorios comúnmente se presentan como: 1) dermatosis urticariana neutrofílica como ocurre en el CAPS; 2) pústulas, abscesos, tractos sinusales o úlceras, como formas sindrómicas de hidradenitis (SAPHO, PAPA, PAPASH o PASH), y 3) diversos tipos de erupciones como paniculitis o livedo reticularis (CANDLE, ADA2).
Las AiD monogénicos son raras, pero existen una serie de enfermedades con una probable base autoinflamatoria poligénica, como es el caso del acné, de la hidradenitis supurativa (HS), la gota o la enfermedad inflamatoria intestinal, así como enfermedades con una clara patogenia mixta (autoinmune y autoinflamatoria), como ocurre en la enfermedad de Behçet o en la psoriasis.
El concepto de enfermedad autoinflamatoria de queratinizaciónEn 2017, Akiyama propuso por primera vez el término AiKD, describiendo características esenciales que las diferenciaban de las AiD. La inflamación se localizará principalmente en la epidermis y en la dermis superior, lo que conduce a la presencia de hiperqueratosis; además, todas estas entidades tendrán factores etiopatogénicos genéticos primarios asociados con la autoinflamación4.
El concepto de la AiKD incluye también enfermedades con autoinflamación y mecanismos autoinmunes mixtos. También ha cambiado el paradigma que se había asumido de que la alteración de la queratinización era el primer evento, y que precedía a la inflamación, como en el acné o en la HS. En la AiKD, la inflamación sería el primer evento, con una queratinización aberrante o defectuosa secundaria. Además, algunas AiKD, como la hidradenitis supurativa, se consideran dermatosis neutrofílicas (ND, por sus siglas en inglés) con una patogenia autoinflamatoria predominante, por lo que los mecanismos patogénicos y las características clínicas de la AiKD y de la ND pueden superponerse.
Mecanismo patogénico y la naturaleza genéticaEn la mayoría de las AiKD se desconocen de manera completa los mecanismos patogénicos, sin embargo, datos nuevos lanzan alguna luz sobre dichos mecanismos (fig. 2)5. Para comenzar, las mutaciones de pérdida de función en el gen IL36RN provocan una regulación positiva de la señalización de la IL-36. Esto conducirá a la secreción de quimiocinas/citocinas por parte de los queratinocitos. La regulación positiva de la vía de señalización de la IL-36 finalmente activará a los neutrófilos y a las células dendríticas, lo que promoverá la polarización de las células Th1 y Th17. Las dermatosis pustulosas relacionadas con el IL-36Ra son la psoriasis pustulosa generalizada (GPP) sin lesiones de psoriasis vulgar, el impétigo herpetiforme y la acrodermatitis continua. Además, algunos estudios han confirmado el papel de la IL-36 en la hidradenitis supurativa6. En segundo lugar, la mutación en el CARD14 hiperactivará el factor nuclear κB (NF-κB; flechas rojas con asteriscos), lo que también conduce a la secreción de quimiocinas/citocinas, IL-36, IL-8, CXCL1, CXCL2 y CCL20, del queratinocito y resultarán en la activación de los neutrófilos y de células dendríticas en la dermis. Así mismo, las células diferenciadas Th1 y Th17, producirán citocinas Th1, como la IL-17. La GPP con psoriasis vulgar y la psoriasis pustulosa palmoplantar son dermatosis psoriasiformes pustulosas mediadas por el CARD14, que presentarán variantes de ganancia en la función del CARD14, como también ocurre en la pitiriasis rubra pilaris (PRP) tipo V. En tercer lugar, se han identificado mutaciones en NLRP1, una proteína sensora del inflamasoma expresada predominantemente en la piel humana, en pacientes con queratosis liquenoide crónica familiar (KLC). El NLRP1 incluye alteraciones en la caspasa-1, una proteína tipo mota asociada a la apoptosis que incluye al CARD (ASC), la IL-1b y la IL-184. De manera similar, en algunos pacientes con hidradenitis supurativa, el rol de la disfunción en el NLRP1 ha sido propuesta como una hipótesis. En cuarto lugar, en pacientes con psoriasis pustulosa grave, como la GPP, las mutaciones en el AP1S3, se han descrito con menor frecuencia. El complejo de proteínas relacionado con el adaptador 1 (AP-1) promueve el tráfico vesicular entre la red trans-Golgi y endosomas. La expresión de AP1S3 estará claramente elevada en los queratinocitos y su silenciamiento interrumpe la translocación endosómica del receptor de reconocimiento de patrón innato TLR-3, lo que da como resultado una marcada inhibición de la regulación de la señalización. Además, su inactivación interrumpe la autofagia de los queratinocitos, lo que lleva a la acumulación anormal de p62, una proteína adaptadora que media la activación de NF-κB, y que da como resultado la señalización positiva de la IL-1 y la sobreexpresión de la IL-36α7,8. En algunos pacientes con HS se han descrito mutaciones funcionales en los genes que codifican la γ-secretasa, incluidos el NCSTN, el PSENEN y el PSEN1. Tales alteraciones genéticas dan como resultado hiperqueratosis, diferenciación disregulada del folículo piloso y formación de quistes a través de la señalización de Notch aberrante. Además, se han demostrado alteraciones en la interleucina 1β, en la interleucina-36, la caspasa-1 y el NLRP3 y una disregulación del eje de las células Th17: Treg en muestras de pacientes con HS9. Finalmente, las mutaciones de pérdida de función en EGFR se han descrito recientemente como una posible causa de hiperqueratosis y autoinflamación, por la regulación positiva de varias redes de respuesta inmunitaria inflamatoria/innata10 (fig. 3).

Mecanismos patogénicos e interacción entre los queratinocitos y las células inmunes en la AiKD. En rojo, enfermedades que se han asociado a todos los mecanismos. GPP: psoriasis pustulosa generalizada; HS: hidradenitis supurativa; KLC: queratosis liquenoide crónica; PRP: pitiriasis rubra pilaris.
La psoriasis pustulosa (fig. 3D) comprende un grupo de trastornos inflamatorios de la piel que pueden clasificarse en GPP, con episodios agudos de formación de pústulas y afectación sistémica; pustulosis palmoplantar (PPP) con erupciones pustulosas crónicas en palmas y plantas; acrodermatitis continua de Hallopeau (ACH) que afecta las puntas de los dedos de manos y pies (fig. 3C); y el impétigo herpetiforme, que es una variante rara de psoriasis pustulosa asociada con el embarazo.
 Queratosis liquenoide crónica; B) Poroqueratosis; C) Acrodermatitis continua de Hallopeau; D) Psoriasis pustulosa.")
En los últimos años, numerosos estudios han descrito la presencia de mutaciones genéticas en la IL36RN, CARD14 y AP1S3. Especialmente, las mutaciones de IL36RN son la anomalía genética más frecuente observada en la psoriasis pustulosa y se han asociado con una edad de presentación más temprana. Se encontraron alelos deletéreos AP1S3 únicamente en el 7 al 10% de los pacientes, y se observaron variantes de CARD14 en un número muy pequeño de sujetos afectados11.
Por otro lado, Twelves et al.8 encontraron una diferencia significativa entre estos subtipos de enfermedades. Específicamente, demostraron que la PPP es más frecuente en mujeres y en pacientes fumadores, con tasas bajas de PV y con características genéticas (baja prevalencia de mutaciones de IL36RN) que son claramente distintas a las observadas en la ACH y en la GPP (con una mayor prevalencia de mutaciones en la IL36RN). Akiyama11, afirma que existe una clara relación entre la GPP sin PV y las mutaciones en IL36RN.
Inicialmente, hubo una controversia en cuanto a si la GPP debería considerarse una AiKD. La mayoría de los autores han propuesto que la GPP sola, puede tratarse como una etiología distinta, porque difiere mucho de la psoriasis vulgar y de la psoriasis pustulosa, en cuanto a las características clínicas, histológicas y genéticas. En cuanto a la presentación clínica y al nivel variable de hiperqueratosis, la diferenciación y proliferación de los queratinocitos se ven frecuentemente afectadas en la epidermis lesional de la GPP. Parece ser una enfermedad poligénica, ya sea por el CARD14 o la IL36RN, dos importantes factores de riesgo para la aparición de GPP12. Recientemente se han encontrado alteraciones en AP1S3 en estos pacientes13,14.
Pitiriasis rubra pilarisLa pitiriasis rubra pilaris (PRP) es una enfermedad inflamatoria de la queratinización, caracterizada por el taponamiento folicular general y la presencia de eritema perifolicular con una configuración característicamente confluente. También se puede observar pitiriasis capitis y queratodermia palmoplantar. Hay 5 subgrupos basados en criterios clínicos. La mayoría son casos esporádicos, excepto en el PRP tipo V (el tipo juvenil atípico), que tiene una expresión clínica diferente y una ocurrencia familiar. Estos pacientes muestran síntomas cutáneos desde la infancia o la primera infancia y suelen tener un curso crónico sin una resolución duradera de la clínica cutánea. Hasta el momento, en el PRP tipo V se han observado mutaciones heterocigóticas de ganancia de función en CARD14, que no se encontraron en otros tipos de PRP. En este sentido, el tipo V podría englobarse como una AiKD5,15. No se excluye que en los otros tipos de PRP exista una base patogénica relacionada con las AiKD, en los que la expresión clínica de la enfermedad implicaría mecanismos patogénicos adicionales o variantes funcionalmente deterioradas del CARD14.
Queratosis liquenoide crónicaLa queratosis liquenoide crónica (KLC) es un trastorno de la queratinización inflamatorio raro, que tendrá una presentación clínica característica, en forma de múltiples pápulas pequeñas con configuraciones confluentes, lineales y reticuladas en el tronco y en las extremidades (fig. 3A). Los pacientes con KLC también pueden tener erupciones similares a la dermatitis seborreica en la cara, queratodermia palmoplantar y deformidades de las uñas. El manejo de la KLC es un desafío, y los tratamientos con retinoides, fototerapia y terapias biológicas han logrado una respuesta variable. Recientemente, se ha descrito que una mutación que conlleve a la ganancia de función en el NLRP1, el gen que codifica para la proteína sensora del inflamasoma NLRP1, está relacionada con la KLC familiar. Por lo tanto, los casos que tenían mutaciones en NLRP1, presentan síntomas de autoinflamación, como son la queratosis folicular cutánea, así como de poliartritis. El NLRP1 es una de las proteínas sensoras del inflamasoma producidas en mayor medida en la piel, así como otros componentes del inflamasoma, como es el caso del CASP1, ASC, IL-1b e IL-18 también se expresarán en los queratinocitos epidérmicos. La mutación responsable de la KLC provoca una activación excesiva del inflamasoma y la secreción de la IL-1, lo que provoca los síntomas inflamatorios y la queratinización. En relación con estos hallazgos, el KLC podría incluirse dentro de las AiKD5,16.
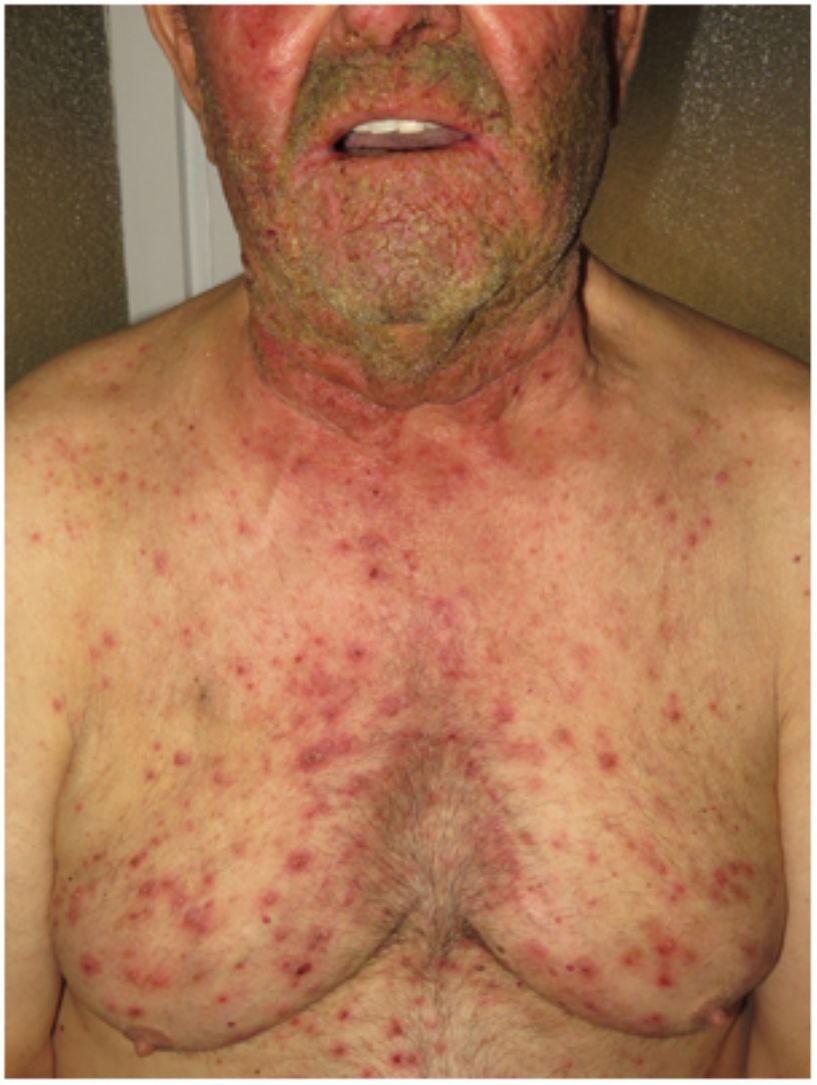
Hidradenitis supurativaLa hidradenitis supurativa (HS) es una enfermedad inflamatoria crónica de la piel caracterizada por la presencia de nódulos, comedones, abscesos, cicatrices hipertróficas y/o tractos sinusales, los que se encuentran típicamente en áreas con abundantes glándulas apocrinas como son: las axilas, las ingles y/o los glúteos9,14,17. Su patogenia incluye la presencia de una queratinización aberrante y de autoinflamación. Se ha demostrado la regulación positiva de la interleucina 1β, la interleucina-36 de la caspasa-1 y el NLRP3 y la disregulación del eje de las células Th17:Treg en muestras provenientes de pacientes con diagnóstico de HS14. La HS podrá coexistir con otras enfermedades autoinflamatorias17. Finalmente, las terapias biológicas como son el adalimumab, el infliximab, el anakinra, el ustekinumab y el secukinumab han demostrado ser eficaces en el tratamiento de la HS moderada a grave. Estos hallazgos en conjunto sugieren que, en aquellos subtipos de HS donde la inflamación precede a la queratinización aberrante, se pueden cumplir los criterios de AiKD propuestos por Akiyama10.
Se han realizado diversos intentos de clasificar a la HS según su patogenia y sus manifestaciones clínicas18. González-Manso et al.19, clasificaron a los pacientes en 2 grupos, que podrían considerarse endotipos, siendo cada uno de ellos la expresión de 2 mecanismos patogénicos diferentes (fig. 4). El «modelo de 2 endotipos» se explica gracias a la secuencia de eventos patogénicos que conducirán a la enfermedad. Sin embargo, la relación existente entre los 2 eventos puede cambiar con el transcurso del tiempo, puede ser bidireccional y los 2 endotipos serán dinámicos. La importancia de estos modelos es que el tratamiento en las primeras etapas de la enfermedad debe de estar orientado al tipo de endotipo.
 y relación con la patogénesis de las AiKD. El endotipo folicular (cluster C1) se caracteriza por tener predominio masculino, tétrada de oclusión folicular y/o acné vulgar, lesiones foliculares-comedónicas que evolucionan a nódulos inflamatorios, con menos abscesos, formaciones de tractos sinusales o cicatrices hipertróficas. Además, la carga genética es mayor con la presencia de mutaciones en la vía de señalización NOTCH/γ-secretasa. Sin embargo, aunque presentan parámetros inflamatorios elevados como el TNF-α sérico y la proteína C reactiva, lo hacen en menor medida en comparación con los del cluster C2. La distribución de la lesión predomina en las zonas posteriores del cuerpo. El endotipo inflamatorio o cluster C2 se caracteriza por tener predominio femenino, la mayoría con sobrepeso u obesidad. Los antecedentes familiares de HS no están tan fuertemente asociados con este grupo, aunque se han descrito mutaciones en MEFV o NLRP3 que promueven la autoinflamación. Se han descrito niveles más altos de inflamación sistémica, especialmente los implicados en la respuesta de las células Th17 (TNF-α, IL-12, IL-23 e IL-17). Las lesiones más típicas son los tractos sinusales y las cicatrices, especialmente en las zonas anteriores del cuerpo. En el endotipo inflamatorio, la autoinflamación precedería a la queratinización, definiéndose como una AiKD sensu stricto. En el endotipo folicular, los pacientes iniciarían el proceso patogénico a través de una alteración de la queratinización que conduce como consecuencia a la autoinflamación.")
Modelo de los 2 endotipos de hidradenitis supurativa (HS) y relación con la patogénesis de las AiKD. El endotipo folicular (cluster C1) se caracteriza por tener predominio masculino, tétrada de oclusión folicular y/o acné vulgar, lesiones foliculares-comedónicas que evolucionan a nódulos inflamatorios, con menos abscesos, formaciones de tractos sinusales o cicatrices hipertróficas. Además, la carga genética es mayor con la presencia de mutaciones en la vía de señalización NOTCH/γ-secretasa. Sin embargo, aunque presentan parámetros inflamatorios elevados como el TNF-α sérico y la proteína C reactiva, lo hacen en menor medida en comparación con los del cluster C2. La distribución de la lesión predomina en las zonas posteriores del cuerpo. El endotipo inflamatorio o cluster C2 se caracteriza por tener predominio femenino, la mayoría con sobrepeso u obesidad. Los antecedentes familiares de HS no están tan fuertemente asociados con este grupo, aunque se han descrito mutaciones en MEFV o NLRP3 que promueven la autoinflamación. Se han descrito niveles más altos de inflamación sistémica, especialmente los implicados en la respuesta de las células Th17 (TNF-α, IL-12, IL-23 e IL-17). Las lesiones más típicas son los tractos sinusales y las cicatrices, especialmente en las zonas anteriores del cuerpo. En el endotipo inflamatorio, la autoinflamación precedería a la queratinización, definiéndose como una AiKD sensu stricto. En el endotipo folicular, los pacientes iniciarían el proceso patogénico a través de una alteración de la queratinización que conduce como consecuencia a la autoinflamación.
Síndrome de queratosis lineal con ictiosis congénita y queratodermia esclerosante
El síndrome de queratosis lineal con ictiosis congénita y queratodermia esclerosante (KLICK, por sus siglas en inglés), es un trastorno cutáneo autosómico recesivo caracterizado por la presencia de queratodermia palmoplantar, placas hiperqueratósicas lineales, descamación ictiosiforme, constricciones circulares alrededor de los dedos y numerosas pápulas distribuidas linealmente en los pliegues de los brazos y de las muñecas20.
Dahlqvist et al.21, encontraron una reducción en los niveles de POMP en algunos pacientes diagnosticados de KLICK, lo que conllevó a una insuficiencia de proteasoma al momento de la diferenciación de los queratinocitos y finalmente desencadenó la autoinflamación. También es interesante señalar que otras alteraciones en el gen POMP dan como resultado enfermedades autoinflamatorias sistémicas, como es el síndrome autoinflamatorio 2 asociado a proteasoma (PRAAS2) y el síndrome de CANDLE. Por tanto, teniendo en cuenta su patogenia, el síndrome de KLICK también cumpliría criterios para ser considerado un AIKD22.
PoroqueratosisLa poroqueratosis se caracteriza por la presencia de máculas o placas hiperqueratósicas, asociadas a un borde con forma de cresta (fig. 3B). Algunos tipos están claramente asociados con la exposición a la luz solar, mientras que en otros una posible relación con diversas enfermedades sistémicas, incluidas una inmunosupresión, una neoplasia y la diabetes23. Los hallazgos histológicos de la poroqueratosis, como la presencia de una columna vertical de paraqueratosis conocida como «lamela cornoide», traduciendo la presencia de una diferenciación y proliferación aberrante de los queratinocitos, asociada a la alteración inflamatoria de la piel, la convierte en una excelente candidata para pertenecer al grupo de las AiKD1,24.
La base genética de esta enfermedad está relacionada con mutaciones en 4 genes de la vía del mevalonato (MVK, MVD, PMVK y FDPS). De hecho, las mutaciones de MVK heterocigotas causan poroqueratosis, y las mutaciones de MVK homocigotas o heterocigotas compuestas dan como resultado hiperinmunoglobulinemia D y síndrome de fiebre periódica, todos ellos parte del espectro de enfermedades autoinflamatorias. Se sabe que la vía del mevalonato produce precursores isoprenoides, por lo que la deficiencia en esa vía provoca la activación del inflamasoma a través de la función anormal de RANKL. En consecuencia, se cree que como resultado causará el crecimiento y la diferenciación anormales de los queratinocitos epidérmicos, así como la autoinflamación en las lesiones de poroqueratosis. Además, en las lesiones cutáneas de poroqueratosis, la expresión de los alelos mutantes es mayor, considerándose un desencadenante de las lesiones. Como mecanismo causal, en la mayoría de los casos se presume de la existencia de un proceso epigenético independiente de metilación del ADN que aún no está del todo claro. Con menor frecuencia, se han reportado alteraciones en la recombinación genómica, lo que resulta en homocigosidad de los alelos mutantes y del ARN aberrante14.
Enfermedades relacionadas con mutaciones en el EGFRLos pacientes con mutaciones de pérdida de función del EGFR presentan descamación cutánea ictiosiforme y una erupción acneiforme. Está clínica se objetiva con frecuencia también en los pacientes que reciben tratamientos oncológicos con inhibidores del EGRF, por lo que esta estaría justificada (fig. 5). Recientemente, Takeichi y Akiyama10, los han incluido en el grupo de las AiKD. Los principales sitios de inflamación estarán en la epidermis y en la dermis superior presentando acantosis, hiperqueratosis, paraqueratosis difusa, infiltración linfocítica perivascular e infiltración de neutrófilos en el epitelio folicular, lo que conduce a una foliculitis. El EGFR mutado regula de manera positiva la fosfolipasa A2, el factor nuclear (NF)-κB y la quinasa c-Jun N-terminal, lo que resulta en la secreción de quimiocinas/citocinas por parte de los queratinocitos y la activación de neutrófilos y células dendríticas en la dermis superficial.
Terapias basadas en los mecanismos patogénicos de la enfermedad autoinflamatoria de la queratinización.")
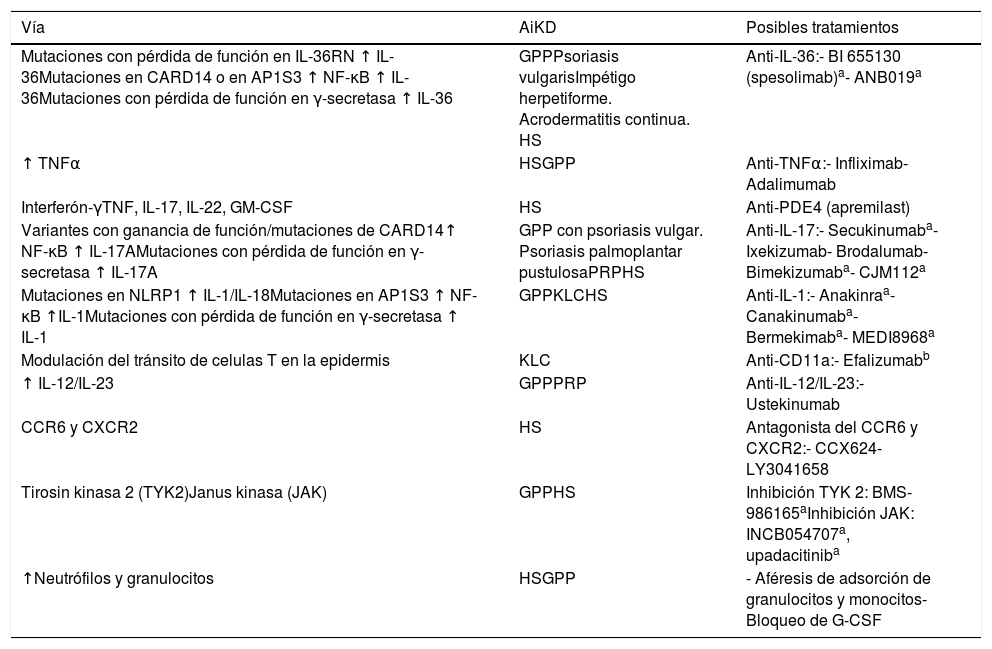
En cuanto a los tratamientos para las AiKD, diversas estrategias terapéuticas se han basado en los mecanismos patogénicos autoinflamatorios descritos, por lo que que se vienen utilizando terapias dirigidas de manera específica contra las moléculas responsables de la alteración en la cascada autoinflamatoria, ya sean las citocinas, sus receptores o las moléculas de señalización. Dichos tratamientos han alcanzado una eficacia prometedora, por lo que se espera que aporten nuevas opciones terapéuticas (tabla 1).
Vías patogénicas relacionadas con AiKD y posibles tratamientos
| Vía | AiKD | Posibles tratamientos |
|---|---|---|
| Mutaciones con pérdida de función en IL-36RN ↑ IL-36Mutaciones en CARD14 o en AP1S3 ↑ NF-κB ↑ IL-36Mutaciones con pérdida de función en γ-secretasa ↑ IL-36 | GPPPsoriasis vulgarisImpétigo herpetiforme. Acrodermatitis continua. HS | Anti-IL-36:- BI 655130 (spesolimab)a- ANB019a |
| ↑ TNFα | HSGPP | Anti-TNFα:- Infliximab- Adalimumab |
| Interferón-γTNF, IL-17, IL-22, GM-CSF | HS | Anti-PDE4 (apremilast) |
| Variantes con ganancia de función/mutaciones de CARD14↑ NF-κB ↑ IL-17AMutaciones con pérdida de función en γ-secretasa ↑ IL-17A | GPP con psoriasis vulgar. Psoriasis palmoplantar pustulosaPRPHS | Anti-IL-17:- Secukinumaba- Ixekizumab- Brodalumab- Bimekizumaba- CJM112a |
| Mutaciones en NLRP1 ↑ IL-1/IL-18Mutaciones en AP1S3 ↑ NF-κB ↑IL-1Mutaciones con pérdida de función en γ-secretasa ↑ IL-1 | GPPKLCHS | Anti-IL-1:- Anakinraa- Canakinumaba- Bermekimaba- MEDI8968a |
| Modulación del tránsito de celulas T en la epidermis | KLC | Anti-CD11a:- Efalizumabb |
| ↑ IL-12/IL-23 | GPPPRP | Anti-IL-12/IL-23:- Ustekinumab |
| CCR6 y CXCR2 | HS | Antagonista del CCR6 y CXCR2:- CCX624- LY3041658 |
| Tirosin kinasa 2 (TYK2)Janus kinasa (JAK) | GPPHS | Inhibición TYK 2: BMS-986165aInhibición JAK: INCB054707a, upadacitiniba |
| ↑Neutrófilos y granulocitos | HSGPP | - Aféresis de adsorción de granulocitos y monocitos- Bloqueo de G-CSF |
AiKD: enfermedad autoinflamatoria de la queratización; GPP: psoriasis pustulosa generalizada, HS: hidradenitis supurativa; KLC: queratosis liquenoide crónica; PRP: pitiriasis rubra pilaris.
En primer lugar, los niveles elevados del TNFα, hacen con que los inhibidores del TNF, como son el infliximab, sean una terapia teóricamente eficaz para el control de la fase aguda de la enfermedad, especialmente de la GPP5. El adalimumab, un anti-TNF, es el único tratamiento biológico aprobado para su uso en la HS.
Por otro lado, la expresión elevada de la IL-17A, ha dado lugar a la realización de diversos ensayos con antagonistas de esta (por ejemplo, el secukinumab, el ixekizumab y el brodalumab), con los que se han obtenido resultados satisfactorios. Cordoro et al.25, reportaron que la inhibición de la IL-17 en un paciente con GPP con deficiencia de IL-36Ra había sido efectiva. La IL-1 de manera similar, participará en la patogenia de las AIKD. Por este motivo, se ha descrito que la terapia con un antagonista del receptor de la IL-1, el anakinra, tuvo éxito en un paciente con GPP causada por mutaciones en la IL36RN y podría ser una opción para los pacientes con KLC y una activación conocida del inflamasoma5. Sorprendentemente, un paciente con diagnóstico de KLC fue resistente a dicho tratamiento, respondiendo sin embargo al efalizumab, un antagonista anti-CD11a que podría actuar modulando el tránsito de las células T hacia la epidermis, con el subsiguiente mal funcionamiento de los queratinocitos26. Desafortunadamente, el efalizumab fue retirado del mercado y ya no está disponible.
Además, los niveles elevados de IL-36 e interferon en la psoriasis vulgar y en la GPP, señalan a estas moléculas como un objetivo potencial. De hecho, algunos ensayos con nuevas moléculas que se encuentran aún en curso, como son el BI 655130 (spesolimab), un anticuerpo monoclonal contra el receptor de la IL-36, han demostrado una reducción en la gravedad de la GPP en fases tempranas. Bachelez et al.27, sugieren que la eficacia del BI 655130, independientemente de la presencia de la mutación en la IL36RN, sugiere que la vía de la IL-36 en sí misma puede jugar un papel importante en su patogénesis. También se está estudiando otro anticuerpo monoclonal humanizado contra el receptor de la IL-36, el ANB019.
Por otro lado, se cree que la IL-12/IL-23 están implicadas en las cascadas inflamatorias que desencadenan la AiKD, incluida la GPP con mutaciones en la IL36RN y en la PRP con mutaciones en el CARD14. En este sentido, la terapia con ustekinumab ha demostrado ser eficaz en estos pacientes28,29. Además, Arakawa et al.30, sugirieron que el tratamiento con acitretina puede apoyar la eficacia del ustekinumab al suprimir las respuestas Th17 a través de los receptores huérfanos relacionados con los retinoides. Esto no es algo nuevo, ya que en fases tempranas la HS se trataba clásicamente con antibióticos, inmunosupresores clásicos y acitretino, reservando los tratamientos biológicos para aquellos pacientes con enfermedad activa y grave. Así mismo, si se tienen en cuenta a los endotipos de la HS, en el caso del endotipo inflamatorio, todos los tratamientos que bloqueen las IL relacionadas con la inhibición de la respuesta Th17, incluidas las citocinas de primera y segunda línea, y los esteroides podrían tener resultados prometedores13. Además, el apremilast, un ihnibidor de la PDE-4, ha demostrado ser eficaz en la enfermedad moderada. Por el contrario, en el endotipo folicular, el acitretino, las tetraciclinas, la dapsona o la colchicina, serán terapias que podrían ser de inicio más útiles19,31,32. Un estudio reciente sobre la progresión histológica de la HS identificó prolongaciones del epitelio, con formación y rotura de quistes. Los tratamientos dirigidos a la inhibición de la formación de estas prolongaciones podrían tener un potencial éxito33.
Además, Campbell et al.34, describieron que el antagonismo de CCR6 y CXCR2, por una pequeña molécula optimizada CCX624, redujo de manera significativa la infiltración de las células T inflamatorias, neutrófilos y células dendríticas en la piel tratada con IL-36a, siendo incluso más eficaz para reducir los síntomas inflamatorios que los anticuerpos monoclonales anti-IL-17RA.
Recientemente, Papp et al.35, objetivaron que la inhibición selectiva de la tirosina quinasa 2 (TYK2) con el agente oral BMS-986165, también podría ser útil para pacientes con GPP.
Los neutrófilos con frecuencia participan en la reacción inflamatoria inducida en las AIKD. De hecho, algunas AiKD se han clasificado como dermatosis neutrofílicas (HS y formas pustulosas de psoriasis); por lo que tratamientos como la dapsona y la colchicina han demostrado tener utilidad en un subconjunto específico de pacientes. Se espera que la aféresis de adsorción de granulocitos y monocitos sea eficaz en algunos casos de AIKD. De hecho, su eficacia ha sido descrita en pacientes con diagnóstico de GPP refractaria a tratamiento36. En relación con lo antes mencionado, la vía del G-CSF es un regulador clave de los neutrófilos, cuya regulación positiva y sus consecuencias parecen ser muy específicas de las lesiones de HS. Por esta razón, bloquear la acción del G-CSF podría representar un nuevo enfoque terapéutico37.
ConclusionesEn los últimos 40 años, los avances en la comprensión de la patogénesis de las dermatosis comunes como es el caso de la psoriasis se han traducido en el desarrollo de terapias dirigidas y altamente efectivas. Hoy en día, el concepto de AiKD, que abarca no solo los trastornos monogénicos raros como el KLC familiar o el PRP de tipo V, sino también las dermatosis comunes con un manejo desafiante como son la HS o la psoriasis pustulosa, será útil para comprender la patogénesis y ser capaces de delinear un manejo terapéutico óptimo de estas entidades. Será obligatorio comprender la interacción que existe entre la inflamación y la queratinización. Los mecanismos de inmunidad innata son, desde la perspectiva darwiniana, una respuesta a lesiones o señales de peligro como patógenos microbianos o estrés, y el descubrimiento de que la inflamación puede conducir a la hiperqueratosis es una consecuencia lógica de la función primaria de esos genes primitivos como potenciadores de la barrera cutánea frente a una agresión externa o interna. El fenómeno de Koebner y la patergia son fenómenos muy conocidos que siguen esta misma línea. Suponemos que las nuevas enfermedades inflamatorias de la queratinización pueden ser reconocidas como AiKD en los próximos años. Los candidatos potenciales serían el liquen plano, o entidades clínicas clásicamente incluidas en el grupo de dermatosis neutrofílicas, como son la pustulosis subcorneal, el pioderma gangrenoso o las manifestaciones cutáneas de la enfermedad inflamatoria intestinal.
Conflicto de interesesEl Dr. Romaní ha recibido honorarios de Abbvie, Novartis, Almirall, Janssen y Leo Pharma por su participación en juntas de asesoramiento, conferencias y como investigador en ensayos clínicos.
El Dr. Peña-Rosado y el Dr. Riera-Martí no tienen ningún conflicto de interés que declarar.



