





Autoinflammatory keratinization disease (AiKD) is a novel clinical concept encompassing diseases with a genetic background and mixed pathogenic mechanisms of autoinflammation and autoimmunity, leading to an aberrant keratinization of the skin. Recent advances in medical genetics have revealed genetic causes and/or predisposing factors for a number of AiKD’s, such as mutations in IL36RN related with pustular psoriasis, acrodermatitis continua and hidradenitis suppurativa, in CARD14 in pityriasis rubra pilaris type V and some forms of pustular psoriasis, and in NLRP1 related with familial keratosis lichenoides chronica (KLC). It is suspected that AiKD pathophysiology would also be involved in non-monogenic disorders.
The bidirectional relationship between inflammation and keratinization should be understood in order to outline optimal management, and new drug development should take both targets into account. We assume that new inflammatory keratinization diseases may be recognized as AiKDs in the coming years.
La enfermedad autoinflamatoria de la queratización (AiKD, de sus siglas en inglés) es un concepto clínico novedoso que engloba a las enfermedades que presentan antecedentes genéticos, así como mecanismos patogénicos mixtos de autoinflamación y autoinmunidad, lo que en su conjunto se traducirá en una queratinización aberrante de la piel. Los recientes avances han revelado causas genéticas y/o factores predisponentes para una serie de AiKD, dentro de los que se pueden enumerar la presencia de las mutaciones en el IL36RN, en relación con la psoriasis pustulosa, la acrodermatitis continua y la hidradenitis supurativa; en el CARD14, en relación con la pitiriasis rubra pilaris tipo V y algunas formas de psoriasis pustulosa, y en el NLRP1 en relación la queratosis liquenoide crónica familiar (KLC, de sus siglas en inglés). Se sospecha que la fisiopatología de la AiKD también estaría presente en algunos trastornos no monogénicos. Se debe de comprender que existe una relación bidireccional entre la inflamación y la queratinización para poder determinar un tratamiento óptimo; así mismo para poder desarrollar nuevos fármacos ambos factores deben de tenerse en cuenta. Probablemente en los próximos años nuevas enfermedades inflamatorias de la queratinización serán incluidas dentro del grupo de las AiKD.
Autoinflammatory keratinization disease (AiKD) is a novel clinical concept that encompasses diseases with a genetic background and mixed pathogenic mechanisms involving autoinflammation and autoimmunity that affect the epidermis and upper dermis, leading to aberrant keratinization of the skin. AiKDs share a key clinical characteristic, namely, they are hyperkeratotic skin lesions that do not respond adequately to standard therapy. The clinical characteristics of the disease are variable. When aberrant keratinization affects the follicular epithelium, the lesions become plugged with keratin and rupture. Correct definition of AiKDs could help us to develop new research lines, especially those analyzing targeted therapy1.
In recent decades, major advances have been made in our understanding of the application of innate and adaptive immunity in dermatologic diseases (Fig. 1). The concept of autoinflammation did not arise until 1999, with the finding of mutations in the germline of the tumor necrosis factor receptor superfamily 1 (TNFRSF1), which is responsible for tumor necrosis receptor–associated periodic syndrome (TRAPS). The term autoinflammation encompasses a group of diseases caused by dysregulation of the innate immune system, which should be differentiated from autoimmune syndromes2.

Some AiKDs are initially thought to be keratinization disorders, such as alteration of arachidonic acid metabolism in psoriasis, thus leading the disease to be treated with oral retinoids. Given the participation of T cells, these diseases were subsequently considered to be autoimmune disorders, and the first biologic therapies were developed to block activation of T cells or cytokines produced by T cells. It was recently postulated that autoinflammation was the main player in the onset of these diseases. The most recent concept in AiKD adds new mechanisms that could drive the development of new targeted therapies. TH indicates helper T cell; TNF, tumor necrosis factor; IL, interleukin.
Innate immunity is based on the defense system, which has traditionally been described as a system that arose earlier in evolution. Therefore, it has a more rapid and nonspecific action and does not leave immunological memory. This system comprises various components: physical barriers, toll-like receptors, phagocytes, natural killer cells, and mastocytes, with complement and antimicrobial peptides being the most important. Innate and adaptive immunity act in a coordinated manner, and most immune disorders are probably a combination of dysfunctions in both systems.
Autoinflammatory Diseases That Affect the SkinInterest in autoinflammatory diseases has grown in the last 10 years. Since they were first defined, more than 30 new genes associated with autoinflammatory diseases affecting different parts of the innate immune system have been identified. The first diseases to be included were hereditary periodic fever syndromes such as familial Mediterranean fever and TRAPS. Other periodic syndromes were soon added, including hereditary hyperimmunoglobulinemia D with periodic fever syndrome (HIDS) and cryopyrin-associated periodic syndrome (CAPS). The concept of autoinflammation subsequently extended to various monogenic diseases, such as Blau syndrome, Majeed syndrome, deficiency of the interleukin 1 receptor antagonist (DIRA), pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA) syndrome, as well as genetic disorders of uncertain origin, such as periodic fever with aphthous stomatitis, pharyngitis, and adenitis (PFAPA) syndrome, Behçet disease, Still disease, Crohn disease, and acquired autoinflammatory syndromes such as Schnitzler syndrome2,3. In clinical terms, autoinflammatory syndromes commonly present as follows: (1) neutrophilic urticarial dermatosis, as in CAPS; (2) pustules, abscesses, sinus tracts, or ulcers, and syndromic forms of hidradenitis (synovitis, acne, pustulosis, hyperostosis, osteitis [SAPHO], PAPA, pyogenic arthritis, pyoderma gangrenosum, acne vulgaris, and HS [PAPASH], and pyoderma gangrenosum, acne, and HS [PASH]); and (3) various types of rash such as panniculitis or livedo reticularis (chronic atypical dermatosis with lipodystrophy and elevated temperature [CANDLE], adenosine deaminase 2 [ADA2] deficiency).
Monogenic autoinflammatory diseases are rare, although some diseases have a probable polygenic autoinflammatory basis, such as acne, hidradenits suppurativa (HS), gout, and inflammatory bowel disease, as well as diseases with a clearly mixed pathogenesis (autoimmune and autoinflammatory), as is the case with Behçet disease and psoriasis.
The Concept of Autoinflammatory Keratinization DiseaseIn 2017, Akiyama et al.4 first proposed the term AiKD to describe the essential characteristics that differentiated these diseases from autoinflammatory diseases. Inflammation mainly affects the epidermis and upper dermis, leading to the presence of hyperkeratosis. In addition, all these diseases have primary genetic etiological and pathogenic factors associated with autoinflammation.
The concept of AiKD also includes diseases with autoinflammation and mixed autoimmune mechanisms. Similarly, a change occurred in the paradigm by which alteration of keratinization was assumed to be the first event and preceded inflammation, as is the case in acne or HS. In AiKD, inflammation is the first event, followed by aberrant, or defective, keratinization. Furthermore, given that some AiKDs, such as HS, were considered neutrophilic dermatoses, with a mainly autoinflammatory origin, the pathogenic mechanisms and clinical characteristics of AiKD and neutrophilic dermatosis might be considered to overlap.
Pathogenic Mechanism and Genetic NatureWhile the pathogenic mechanisms of most AiKDs are not completely known, new data shed light on some of them (Fig. 2)5. First, loss-of-function mutations in IL36RN lead to positive regulation of IL-36 signaling. This in turn leads to secretion of chemokines/cytokines by keratinocytes. Positive regulation of the IL-36 signaling pathway eventually activates neutrophils and dendritic cells, thus leading to polarization of type 1 helper T cells (TH1) and TH17 cells. The pustular dermatoses associated with IL-36Ra are generalized pustular psoriasis (GPP) without psoriasis vulgaris lesions, impetigo herpetiformis, and acrodermatitis continua. Furthermore, some studies have confirmed the role of IL-36 in HS6. Second, the mutation in CARD14 hyperactivates nuclear factor κB (NF-κB; red arrows with asterisks), also leading to secretion of chemokines and cytokines—IL-36, IL-8, CXCL1, CXCL2, CCL20—in the keratinocyte and activation of neutrophils and dendritic cells in the dermis. Similarly, differentiated TH1 and TH17 cells produce TH1 cytokines, such as IL-17. GPP with psoriasis vulgaris and palmoplantar pustular psoriasis are pustular psoriasiform dermatoses mediated by CARD14 that present gain-of-function variants in CARD14, as is also the case in pityriasis rubra pilaris (PRP) type V. Third, mutations have been identified in NLRP1, an inflammasome sensor expressed mainly in human skin in patients with familial keratosis lichenoides chronica (KLC). NLRP1 includes alterations in caspase 1, an apoptosis-associated speck-like protein containing a CARD (ASC), IL-1β, and IL-184. Similarly, in some patients with HS, a role for NLRP1 dysfunction has been hypothesized. Fourth, in the case of patients with severe pustular psoriasis, such as GPP, mutations in AP1S3 have been less frequently reported. Adaptor protein complex 1 (AP-1) promotes vesicular trafficking between the trans-Golgi network and endosomes. Expression of AP1S3 is clearly elevated in keratinocytes, and its silencing interrupts endosomal translocation of the innate pattern recognition receptor TLR-3, resulting in markedly inhibited regulation of signaling. Furthermore, its inactivation interrupts keratinocyte autophagy, leading to abnormal accumulation of p62, an adaptor protein that mediates activation of NF-κB and results in positive signaling of IL-1 and overexpression of IL-36α7,8. In some patients with HS, functional mutations have been reported in genes encoding γ-secretase, including NCSTN, PSENEN, and PSEN1. These genetic abnormalities result in hyperkeratosis, dysregulated differentiation of the hair follicle, and formation of cysts via signaling of aberrant Notch. Furthermore, abnormalities have been shown in IL-1β, IL-36, caspase-1, and NLRP3, as has dysregulation of the TH17-Treg axis in samples from patients with HS9. Finally, loss of function mutations in EGFR were recently reported to be a potential cause of hyperkeratosis and autoinflammation owing to positive regulation of various inflammatory/innate immune response networks (Fig. 3)10.


Pustular psoriasis (Fig. 3) encompasses a group of inflammatory disorders of the skin that can be classified as follows: GPP, with acute episodes involving formation of pustules and systemic involvement; pustulosis palmoplantaris (PPP), with chronic pustular eruptions on the palms and soles; acrodermatitis continua of Hallopeau (ACH), which affects the tips of the fingers and toes (Fig. 3C); and impetigo herpetiformis, which is a rare variant of pustular psoriasis of pregnancy.
In recent years, several studies have reported the presence of mutations in the IL36RN, CARD14, and AP1S3 genes. Mutations in IL36RN are the most frequent genetic abnormality in pustular psoriasis and have been associated with earlier age at onset. Deleterious AP1S3 alleles were found only in 7% to 10% of patients, and variants in CARD14 were observed in a very small number of affected persons11.
Twelves et al.8 found a significant difference between these disease subtypes. Specifically, they showed that PPP is more frequent in women and smokers, with low rates of psoriasis vulgaris and genetic characteristics (low prevalence of mutations in IL36RN) that clearly differ from those observed in ACH and GPP (with a greater prevalence of mutations in IL36RN). Akiyama11 reported a clear association between GPP without psoriasis vulgaris and mutations in IL36RN.
Initially, there was some controversy over whether GPP should be considered an AiKD. Most authors proposed that GPP alone could be treated as a distinct etiology, because it differs considerably from psoriasis vulgaris and pustular psoriasis in terms of clinical, histopathologic, and genetic characteristics. As for clinical presentation and variable level of hyperkeratosis, keratinocyte differentiation and proliferation are often affected in lesional epidermis in GPP. It seems to be a polygenic disease, caused either by CARD14 or IL36RN, which are both major risk factors for GPP12. Alterations in AP1S3 were recently reported in affected patients13,14.
Pityriasis rubra pilarisPityriasis rubra pilaris (PRP) is an inflammatory keratinization disease that is characterized by generalized follicular plugging and perifollicular erythema with a typical coalescent arrangement. Pityriasis capitis and palmoplantar keratoderma can also be observed. Based on clinical criteria, we can identify 5 subgroups. Most involve sporadic cases, except for PRP type V (atypical juvenile-onset), which is familial and whose clinical expression differs from the other types. These patients experience cutaneous symptoms from childhood or early infancy, and their disease course is chronic, with no lasting resolution of the cutaneous symptoms. Heterozygous gain-of-function mutations have been observed in CARD14 in patients with PRP type V: these have not been reported in other types of PRP. In this sense, type V could be considered an AiKD5,15. We cannot rule out the possibility that in other types of PRP, pathogenesis is associated with AiKD, where clinical expression of the disease would involve additional pathogenic mechanisms or functionally impaired variants in CARD14.
Keratosis lichenoides chronicaKLC is a rare inflammatory keratinization disease with a characteristic clinical presentation that takes the form of coalescent multiple small papules arranged in a linear or reticulate pattern on the trunk and extremities (Fig. 3A). Patients with KLC may also experience rash similar to seborrheic dermatitis on the face, palmoplantar keratoderma, and nail involvement. Management of KLC is challenging, and treatment with retinoids, phototherapy, and biologics has led to varying responses. A recently described gain-of-function mutation in NLRP1, the gene that codes for the inflammasome sensor NLRP1, is associated with familial KLC. Therefore, patients with mutations in NLRP1 present with symptoms of autoinflammation, such as keratosis pilaris and polyarthritis. The protein NLRP1 is an inflammasome sensor produced mainly in the skin, as are other components of the inflammasome, for example, CASP1, ASC, IL-1β, and IL-18, which are also expressed in epidermal keratinocytes. The mutation responsible for KLC leads to excessive activation of the inflammasome and secretion of IL-1, thus triggering inflammatory symptoms and keratinization. Given these findings, KLC could be considered an AiKD5,16.
Hidradenitis suppurativaHS is a chronic inflammatory skin disease characterized by the presence of nodules, comedones, abscesses, hypertrophic scars, and/or sinus tracts, which are typically found in areas rich in apocrine glands such as the axillas, groin, and gluteus muscles9,14,17. Pathogenesis includes aberrant keratinization and autoinflammation. There have been reports of positive regulation of IL-1β, IL-36, caspase-1, and NLRP3 and dysregulation of the TH17-Treg axis in samples from patients diagnosed with HS14. HS can co-occur with other autoinflammatory diseases17. Finally, biologics such as adalimumab, infliximab, anakinra, ustekinumab, and secukinumab have proven effective for the treatment of moderate-to-severe HS. Taken together, these findings suggest that, in subtypes of HS where inflammation precedes aberrant keratinization, the criteria for AiKD proposed by Takeichi and Akiyama10 are fulfilled.
Various attempts have been made to classify HS by pathogenesis and clinical manifestations18. González-Manso et al.19 classified patients into 2 groups that could be considered endotypes, with each representing the expression of 2 separate pathogenic mechanisms (Fig. 4). The “2-endotype model” can be explained by the sequence of pathogenic events that lead to the disease. However, the association between the 2 events can change over time and may be bidirectional, and the 2 endotypes can be dynamic. The importance of these models is that treatment of the early stages of the disease should target the endotype.
 and their association with the pathogenesis of autoinflammatory keratinization disease (AiKD). The follicular endotype (cluster C1) is affected by male predominance, follicular occlusion tetrad and/or acne vulgaris, follicular-comedone-like lesions that progress to inflammatory nodules, with fewer abscesses, sinus tracts, and hypertrophic scars. Furthermore, the genetic burden is greater, with the presence of mutations in the Notch/γ-secretase signaling pathway. However, while they present elevated inflammatory parameters, such as serum TNF-α and C-reactive protein, they do so to a lesser extent than those of the C2 cluster. The lesion mainly affects the posterior areas of the body. The inflammatory endotype, or C2 cluster, is more common in women, mainly in those who have obesity or overweight. A family history of HS is not as closely associated with this group, although mutations in MEFV and NLRP3 that promote inflammation have been reported. Higher levels of systemic inflammation have been reported in the TH17 response (TNF-α, IL-12, IL-23, and IL-17). The most typical lesions are sinus tracts and scars, especially on the anterior areas of the body. In the inflammatory endotype, autoinflammation precedes keratinization and is thus defined as an AiKD sensu stricto. In the follicular endotype, pathogenesis begins through an alteration of keratinization that consequently leads to autoinflammation.")
Model of the 2 endotypes of hidradenitis suppurativa (HS) and their association with the pathogenesis of autoinflammatory keratinization disease (AiKD). The follicular endotype (cluster C1) is affected by male predominance, follicular occlusion tetrad and/or acne vulgaris, follicular-comedone-like lesions that progress to inflammatory nodules, with fewer abscesses, sinus tracts, and hypertrophic scars. Furthermore, the genetic burden is greater, with the presence of mutations in the Notch/γ-secretase signaling pathway. However, while they present elevated inflammatory parameters, such as serum TNF-α and C-reactive protein, they do so to a lesser extent than those of the C2 cluster. The lesion mainly affects the posterior areas of the body. The inflammatory endotype, or C2 cluster, is more common in women, mainly in those who have obesity or overweight. A family history of HS is not as closely associated with this group, although mutations in MEFV and NLRP3 that promote inflammation have been reported. Higher levels of systemic inflammation have been reported in the TH17 response (TNF-α, IL-12, IL-23, and IL-17). The most typical lesions are sinus tracts and scars, especially on the anterior areas of the body. In the inflammatory endotype, autoinflammation precedes keratinization and is thus defined as an AiKD sensu stricto. In the follicular endotype, pathogenesis begins through an alteration of keratinization that consequently leads to autoinflammation.
Keratosis linearis with ichthyosis congenita and sclerosing keratoderma (KLICK) syndrome is an autosomal recessive skin disease characterized by the presence of palmoplantar keratoderma, linear hyperkeratotic plaques, ichthyosiform scaling, circular constrictions around the fingers, and multiple papules arranged linearly in the arm folds and on the wrists20.
Dahlqvist et al.21 found reduced levels of proteasome maturation protein (POMP) in some patients diagnosed with KLICK syndrome, leading to insufficient proteasome on differentiation of the keratinocytes and, eventually, triggering autoinflammation. It is also noteworthy that other alterations in the POMP gene result in systemic autoinflammatory diseases, such as proteasome-associated autoinflammatory syndrome 2 (PRAAS2) and CANDLE syndrome. Therefore, taking its pathogenesis into account, KLICK syndrome would also fulfill the criteria for AiKD22.
PorokeratosisPorokeratosis is characterized by the presence of hyperkeratotic plaques or macules with a ridge-like border (Fig. 3B). Some types are clearly associated with exposure to sunlight, whereas others are potentially associated with various systemic diseases, including immunosuppression, cancer, and diabetes23. Histological findings, namely, presence of a vertical parakeratotic column known as the cornoid lamella, which indicates aberrant proliferation and differentiation of keratinocytes associated with inflammation of the skin, make porokeratosis an excellent candidate for AiKD1,24.
The underlying genetic basis for this disease involves mutations in 4 genes of the mevalonate pathway (MVK, MVD, PMVK, and FDPS). In fact, heterozygous mutations in MVK cause porokeratosis, and compound homozygous or heterozygous mutations in MVK lead to hyperimmunoglobulinemia D and periodic fever syndrome, all of which lie on the spectrum of autoinflammatory diseases. The mevalonate pathway is known to produce isoprenoid precursors; therefore, a deficiency on this pathway triggers activation of the inflammasome via abnormal functioning of RANKL. This is in turn thought to result in abnormal growth and differentiation of epidermal keratinocytes, as well as autoinflammation in porokeratosis lesions. Furthermore, in the skin lesions of porokeratosis, the level of expression of mutant alleles is higher, thus leading it to be considered a trigger of the lesions. In most cases, the underlying mechanism is thought to be an independent epigenetic DNA methylation process, which has not yet been fully elucidated. Less frequently, there have been reports of abnormalities in genomic recombination, resulting in homozygosity of mutant alleles and aberrant DNA14.
Diseases associated with mutations in EGFRPatients with loss-of-function mutations in EGFR present with ichthyosiform scaling and acneiform eruption. These clinical findings are also commonly observed in patients who receive EGFR inhibitors for treatment of cancer; therefore, inclusion of these diseases would be justified (Fig. 5). Takeichi and Akiyama10 recently included them in AiKDs. The main sites of inflammation are the epidermis and upper dermis, with acanthosis, hyperkeratosis, diffuse parakeratosis, perivascular lymphocytic infiltration, and infiltration of the follicular epithelium by neutrophils, leading to folliculitis. Mutated EGFR upregulates phospholipase A2, NF-κB, and c-Jun N-terminal kinase, resulting in secretion of chemokines/cytokines by keratinocytes and activation of neutrophils and dendritic cells in the superficial dermis.
Therapies Based on the Pathogenic Mechanisms of Autoinflammatory Keratinization Disease
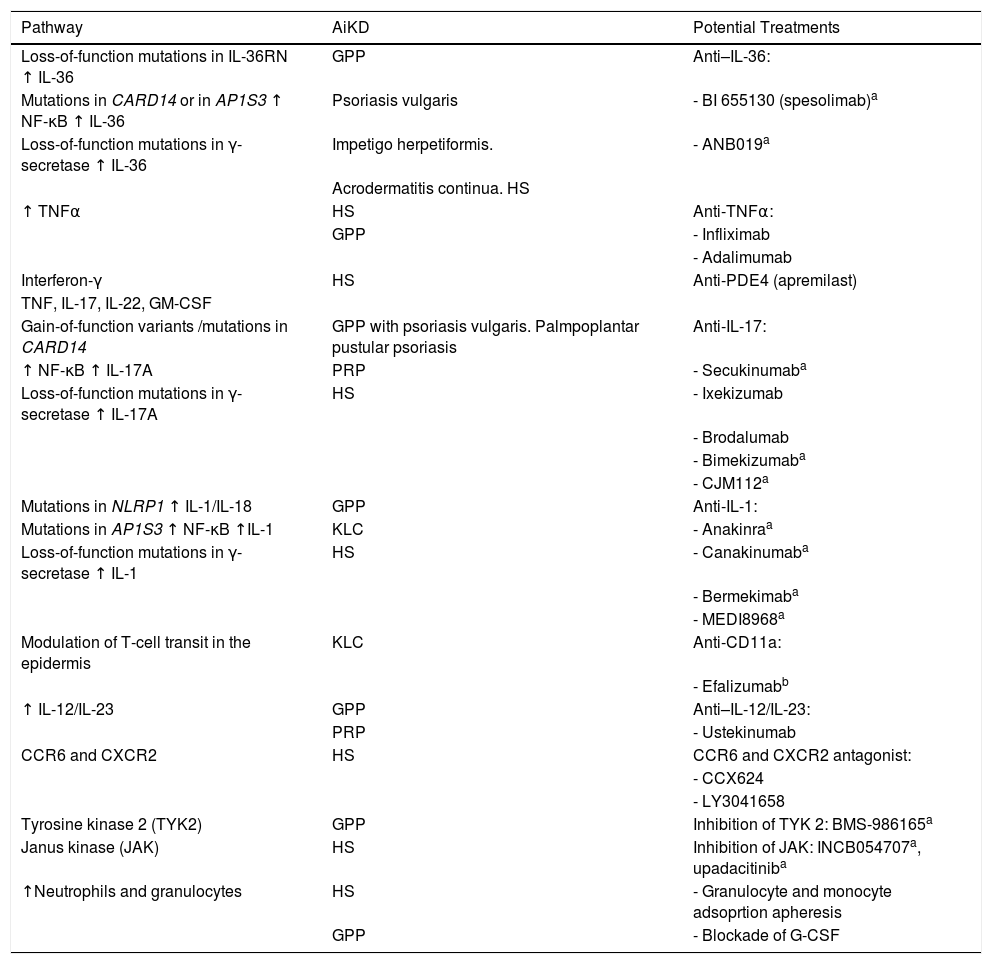
Various strategies for the treatment of AiKDs have been based on the underlying autoinflammatory pathogenic mechanisms. Therefore, the approach used comprises treatments targeting the molecules responsible for alteration of the autoinflammatory cascade, namely, cytokines, their receptors, and signaling molecules. Given that the effectiveness of these treatments is promising, they are expected to generate new therapeutic options (Table 1).
Pathogenic pathways associated with AiKD and potential treatments.
| Pathway | AiKD | Potential Treatments |
|---|---|---|
| Loss-of-function mutations in IL-36RN ↑ IL-36 | GPP | Anti–IL-36: |
| Mutations in CARD14 or in AP1S3 ↑ NF-κB ↑ IL-36 | Psoriasis vulgaris | - BI 655130 (spesolimab)a |
| Loss-of-function mutations in γ-secretase ↑ IL-36 | Impetigo herpetiformis. | - ANB019a |
| Acrodermatitis continua. HS | ||
| ↑ TNFα | HS | Anti-TNFα: |
| GPP | - Infliximab | |
| - Adalimumab | ||
| Interferon-γ | HS | Anti-PDE4 (apremilast) |
| TNF, IL-17, IL-22, GM-CSF | ||
| Gain-of-function variants /mutations in CARD14 | GPP with psoriasis vulgaris. Palmpoplantar pustular psoriasis | Anti-IL-17: |
| ↑ NF-κB ↑ IL-17A | PRP | - Secukinumaba |
| Loss-of-function mutations in γ-secretase ↑ IL-17A | HS | - Ixekizumab |
| - Brodalumab | ||
| - Bimekizumaba | ||
| - CJM112a | ||
| Mutations in NLRP1 ↑ IL-1/IL-18 | GPP | Anti-IL-1: |
| Mutations in AP1S3 ↑ NF-κB ↑IL-1 | KLC | - Anakinraa |
| Loss-of-function mutations in γ-secretase ↑ IL-1 | HS | - Canakinumaba |
| - Bermekimaba | ||
| - MEDI8968a | ||
| Modulation of T-cell transit in the epidermis | KLC | Anti-CD11a: |
| - Efalizumabb | ||
| ↑ IL-12/IL-23 | GPP | Anti–IL-12/IL-23: |
| PRP | - Ustekinumab | |
| CCR6 and CXCR2 | HS | CCR6 and CXCR2 antagonist: |
| - CCX624 | ||
| - LY3041658 | ||
| Tyrosine kinase 2 (TYK2) | GPP | Inhibition of TYK 2: BMS-986165a |
| Janus kinase (JAK) | HS | Inhibition of JAK: INCB054707a, upadacitiniba |
| ↑Neutrophils and granulocytes | HS | - Granulocyte and monocyte adsoprtion apheresis |
| GPP | - Blockade of G-CSF |
Abbreviations: AiKD, autoinflammatory keratinization disease; G-CSF, granulocyte colony-stimulating factor; GPP, generalized pustular psoriasis; HS, hidradenitis suppurativa; KLC, keratosis lichenoides chronica; PRP, pityriasis rubra pilaris.
In the first place, high levels of TNF-α make TNF inhibitors such as infliximab theoretically efficacious for control of the acute phase of the disease, especially in the case of GPP5. The anti-TNF agent adalimumab is the only biologic approved for HS.
Furthermore, the increased expression of IL-17A observed led to various trials with IL-17A antagonists (e.g., secukinumab, ixekizumab, and brodalumab), for which findings have been satisfactory. Cordoro et al.25 reported that inhibition of IL-17 in a patient with GPP and IL-36Ra deficiency had proven effective. Similarly, given that IL-1 participates in the pathogenesis of AiKD, therapy with an IL-1 receptor antagonist, anakinra, was successful in a patient with GPP caused by mutations in IL36RN and could be an option in patients with KLC and known activation of the inflammasome5. Surprisingly, a patient diagnosed with KLC did not respond to this approach, yet responded to efalizumab, an anti-CD11a antagonist that could act by modulating transit of T cells to the epidermis, with subsequent keratinocyte malfunction26. Unfortunately, efalizumab was withdrawn from the market and is no longer available.
High IL-36 and interferon levels in psoriasis vulgaris and GPP point to these molecules as a potential target. In fact, ongoing trials with new drugs, such as BI 655130 (spesolimab), a monoclonal antibody against the IL-36 receptor, have reduced the severity of GPP in the early phases. According to Bachelez et al.27, the efficacy of BI 655130 suggests that the IL-36 pathway itself could play a key role in pathogenesis, independently of the mutation in IL36RN. Also under study is ANB019, another humanized monoclonal antibody targeting the IL-36 receptor.
IL-12 and IL-23 are thought to be involved in the inflammatory cascades that trigger AiKD, including GPP (with mutations in IL36RN) and in PRP (with mutations in CARD14). In this sense, ustekinumab has proven efficacious in affected patients28,29. Arakawa et al.30 suggested that treatment with acitretin could support the efficacy of ustekinumab by suppressing the TH17 response via retinoid-related orphan receptors. This finding is not novel, since the early phases of HS were traditionally treated with antibiotics, classic immunosuppressants, and acitretin, with biologics being reserved for patients with active, severe disease. Similarly, if the endotypes of HS are considered, in the case of the inflammatory endotype, all interleukin blockers associated with inhibition of the TH17 response (including first- and second-line cytokines) and steroids could yield promising results13. Moreover, apremilast, a PDE-4 inhibitor, has proven effective in moderate disease. In contrast, with the follicular endotype, acitretin, tetracyclines, dapsone, and colchicine could initially prove more useful19,31,32. A recent study on the histologic progression of HS revealed epithelial tendrils, with formation and rupture of cysts. Treatments that inhibit the formation of these tendrils could prove successful33.
Campbell et al.34 reported that an optimized small-molecule antagonist, CCX624, targeting CCR6 and CXCR2 significantly diminished inflammatory T-cell, neutrophil, and dendritic cell infiltrates in skin treated with IL-36a and even proved to be more effective for reducing inflammatory symptoms than anti-IL-17RA monoclonal antibodies.
Papp et al.35 found that selective inhibition of tyrosine kinase 2 with the oral agent BMS-986165 could also prove useful in patients with GPP.
Neutrophils frequently participate in the inflammatory reaction induced in AiKDs, and some AiKDs have been classified as neutrophilic dermatoses (HS and pustular forms of psoriasis). Therefore, treatments such as dapsone and colchicine have proven useful in a specific subgroup of patients. Granulocyte and monocyte adsorption apheresis is expected to be effective in some cases of AiKD; in fact, it has been reported to be effective in patients diagnosed with refractory GPP36. Accordingly, the granulocyte colony-stimulating factor pathway is a key regulator of neutrophils, whose positive regulation and its effects seem to be very specific for HS lesions. Consequently, blocking the action of granulocyte colony-stimulating factor could prove to be a new therapeutic approach37.
ConclusionsDuring the last 40 years, advances in our understanding of the pathogenesis of common skin diseases such a psoriasis have translated into highly effective targeted therapies. Today, the concept of AiKD, which encompasses not only rare monogenic disorders, such as familial KLC and PRP type V, but also common skin conditions that are challenging to manage (e.g., HS or pustular psoriasis), can help us understand pathogenesis and enable us to design optimal therapies for these diseases. An understanding of the interaction between inflammation and keratinization is mandatory. From a darwinian perspective, the mechanisms underlying innate immunity are a response to lesions or danger signals, such as microbial pathogens or stress. In addition, the discovery that inflammation can lead to hyperkeratosis is a logical consequence of the primary function of these primitive genes as boosters of the skin barrier against an internal or external insult. Koebner phenomenon and pathergy are well known phenomena that follow this same line. We believe that new inflammatory keratinization diseases will be considered AiKDs in the coming years. Potential candidates include lichen planus and conditions traditionally considered neutrophilic dermatoses, such as subcorneal pustular dermatosis, pyoderma gangrenosum, and cutaneous manifestations of inflammatory bowel disease.
Conflicts of interestDr. Romaní has received fees from AbbVie, Novartis, Almirall, Janssen, and Leo Pharma for participating in advisory boards and talks and for participating as a researcher in clinical trials.
Dr. Peña-Rosado and Dr. Riera-Martí declare that they have no conflicts of interest.
Please cite this article as: Peña-Rosado A, Riera-Martí N, Expósito-Serrano V, Romaní J. Trastornos autoinflamatorios de la queratinización. Actas Dermosifiliogr. 2021;112:891–900.


