









El sarcoma de Kaposi es un sarcoma vascular con cuatro variantes clínicas: el clásico, que asienta preferentemente en las extremidades de pacientes ancianos, de curso crónico y poco agresivo; el endémico de África central; el de pacientes inmunodeprimidos, y el asociado a SIDA. En todas las variedades se ha demostrado que el virus herpes tipo8 es el agente etiológico. El angiosarcoma cutáneo es una de las neoplasias cutáneas de peor pronóstico, con gran tendencia a la recidiva local y una supervivencia a 5años del 10-50%. Existen 3 grandes variedades de angiosarcomas cutáneos: los idiopáticos de cara y cuero cabelludo, los desarrollados sobre áreas de linfedema crónico y los que aparecen sobre áreas de piel irradiada. El único tratamiento potencialmente curativo es la cirugía asociada o no a radioterapia, pero su mala delimitación y su carácter multicéntrico obligan en muchos casos a emplear tratamientos paliativos con quimio y/o radioterapia.
Kaposi sarcoma is a vascular sarcoma with 4 clinical variants: classic Kaposi sarcoma, which mainly affect the extremities of elderly patients and follows a chronic, generally indolent course; African Kaposi sarcoma; immunosuppression-associated Kaposi sarcoma; and AIDS-associated Kaposi sarcoma. Type8 human herpesvirus is the etiologic agent in all 4variants. Cutaneous angiosarcoma is a cutaneous neoplasm with a very poor prognosis. It carries a high probability of local relapse and has a 10% to 15% survival rate at 5years. There are 3 main variants of cutaneous angiosarcoma: idiopathic angiosarcoma of the face and scalp; Stewart-Treves syndrome; and postradiation angiosarcoma. The only potentially curative treatment is surgery with or without radiotherapy. However, its indistinct borders and multicentric nature mean that treatment is often palliative with chemotherapy, radiotherapy, or both.
El sarcoma de Kaposi (SK) es un tumor angioproliferativo asociado a la infección por virus herpes humano tipo8 (VHH-8)1,2. Se describen 4 variantes (tabla 1):
Epidemiología y características clínicas de los distintos subtipos de sarcoma de Kaposi
| SK clásico | SK endémico | SK asociado a VIH | SK asociado a inmunosupresión | |
|---|---|---|---|---|
| Edad | >60 años | 30-45 años | 20-50 años | <60 años |
| Grupos de riesgo | Origen mediterráneo y judíos | Población africana | Infectados por VIH, HSH | Trasplantados, enfermedades autoinmunes |
| Localización | Miembros inferiores | Miembros inferiores | Cefálico, oral, visceral | Extremidades |
| Manifestaciones extracutáneas | Infrecuentes | Frecuentemente linfoadenopático | Frecuentes | Posibles |
| Evolución | Curso indolente | Progresivo | Agresivo. Puede regresar con antirretrovirales | Variable, puede regresar con disminución de la inmunosupresión |
HSH: hombres que tiene sexo con hombres; SK: sarcoma de Kaposi; VIH: virus de la inmunodeficiencia humana.
Sarcoma de Kaposi clásico. Es un tumor infrecuente que afecta a hombres1,3 del área mediterránea o centroeuropea, con una incidencia entre 0,18-13,2 casos/1064, siendo más frecuente en hombres con edema crónico de piernas, diabetes mellitus y usuarios de corticoides. Se presenta como placas o nódulos eritematovioláceos, únicos o múltiples, de crecimiento lento, asociado raramente a linfedema de extremidades y afectación gastrointestinal y ganglionar. El curso clínico es indolente, y un 2% de los pacientes fallecen por enfermedad diseminada.
Sarcoma de Kaposi endémico. Descrito en África ecuatorial, afecta a adultos jóvenes o prepúberes. En los adultos sigue un curso indolente o agresivo con afectación de tejido subcutáneo y óseo, mientras que en los niños se manifiesta agresivo con linfoadenopatías generalizadas, compromiso visceral y ausencia de (o escasas) lesiones cutáneas5.
Sarcoma de Kaposi asociado a inmunosupresión (o iatrogénico). Descrito en pacientes en tratamiento inmunosupresor, preferentemente trasplantados. El riesgo de desarrollar SK se estima entre 150 a 200 veces el de la población general, con un tiempo promedio de 18meses6.
Sarcoma de Kaposi asociado a VIH (o epidémico). Descrito, en hombres que tienen sexo con hombres (HSH)-VIH+. Previo a la terapia antirretroviral de gran actividad (TARGA) se calculaba que un 25% de los HSH-VIH+ desarrollarían un SK, porcentaje que ha disminuido progresivamente2,7 (figs. 1 y 2). En estos pacientes puede afectar la piel y mucosas, ganglios linfáticos, tracto gastrointestinal, pulmones, bazo e hígado7. También se ha documentado SK en HSH-VIH−, de curso indolente8.


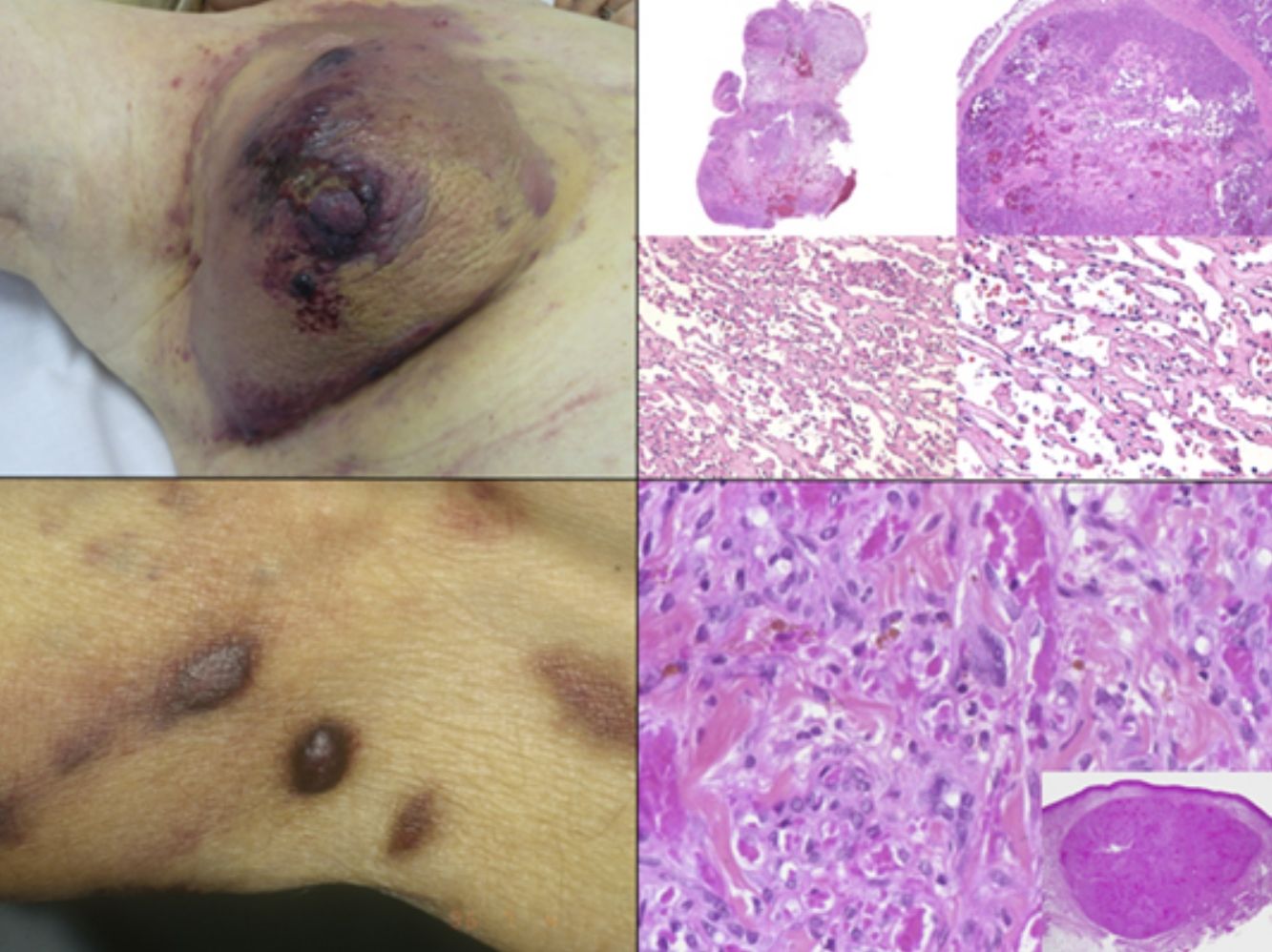
El diagnóstico es clínico, pero es recomendable confirmarlo por biopsia. En el examen anatomopatológico se observan en todo el grosor de la dermis células fusiformes, proliferación de vasos irregulares con hendiduras vasculares, con extravasación de hematíes, e infiltrado leucoplasmocitario y glóbulos hialinos intra y extracelulares, así como el denominado signo del promontorio (fig. 3). La PCR y la tinción inmunohistoquímica para la detección del antígeno nuclear asociado a latencia (LANA-1) del virus VHH-8 son positivos1.
 Visión panorámica de sarcoma de Kaposi en fase nodular. Nódulo bien circunscrito en dermis. b) Lesión densamente celular con algún espacio en forma de grieta. c) Detalle de las células fusiformes y de los hematíes en el interior de los pequeños vasos. d) Positividad nuclear para la inmunohistoquímica VHH-8 específica del sarcoma de Kaposi.")
Histología del sarcoma de Kaposi.
a) Visión panorámica de sarcoma de Kaposi en fase nodular. Nódulo bien circunscrito en dermis.
b) Lesión densamente celular con algún espacio en forma de grieta.
c) Detalle de las células fusiformes y de los hematíes en el interior de los pequeños vasos.
d) Positividad nuclear para la inmunohistoquímica VHH-8 específica del sarcoma de Kaposi.
Sarcoma de Kaposi clásico. Dada la clínica (edad, afectación local, infrecuente afectación visceral y curso indolente), es suficiente la exploración cutánea y ganglionar. Las pruebas complementarias se realizan si el paciente presenta síntomas de compromiso visceral5.
Sarcoma de Kaposi asociado a inmunosupresión. Al igual que en el SK asociado a VIH, no existe una estadificación consensuada para el SK asociado a inmunosupresión, por lo que las pruebas y los criterios de aplicación que se recomiendan son habitualmente los mismos que en el SK asociado a VIH.
Sarcoma de Kaposi asociado a VIH. No existe un sistema de estadificación aceptado. Se recomienda realizar una radiografía de tórax y, en caso de detectar alteraciones sugerentes de compromiso pulmonar, una broncoscopia o una tomografía computarizada de tórax. También se aconseja descartar sangre oculta en heces y, si es positiva, realizar una endoscopia digestiva.
El AIDS Clinical Trials Group Oncology Committee propuso en 1989 una estadificación (tabla 2) basada en la extensión de la enfermedad, recuento de linfocitosT CD4 y compromiso sistémico (enfermedades oportunistas, síntomasB como fiebre, pérdida de peso o diarrea persistente, y rendimiento menor a 70puntos de la escala de Karnofsky). Un análisis prospectivo mostró que estas variables se relacionaban con la supervivencia de los pacientes, siendo factores de buen pronóstico la enfermedad limitada, los linfocitos CD4 superiores a 150células/mm3 y la ausencia de compromiso sistémico9.
Estadificación propuesta por el AIDS Clinical Trials Group Oncology Committee
| Buen pronóstico (0) | Mal pronóstico (1) | |
|---|---|---|
| Tumor | Tumor confinado a la piel o a ganglios o mínima afectación oral | Tumor asociado a edema o ulceración Afectación oral grave SK digestivo SK visceral (no ganglionar) |
| Inmunidad | CD4 ≥ 200/μl | CD4 <200/μl |
| Enfermedad sistémica | Sin enfermedades oportunistas o muguet Sin síntomas B Índice de Karnofsky ≥ 70 | Infecciones oportunistas o muguet Síntomas Ba Indice de Karnofsky <70 Otras enfermedades relacionadas con el VIH (p.ej., enfermedad neurológica o linfoma) |
SK: sarcoma de Kaposi; VIH: virus de la inmunodeficiencia humana.
Estas propuestas de estadificación se realizaron antes de la TARGA y no incluyen la carga vírica, por lo que la aplicación de las estatificaciones ha quedado relegada a los estudios clínicos.
TratamientoSarcoma de Kaposi clásicoExisten pocos ensayos clínicos comparativos entre los diferentes tratamientos del SK clásico. Se emplean habitualmente los mismos fármacos y pautas que los aplicados en el SK epidémico (tabla 3).
- 1.
Lesiones únicas.
- •
Observación clínica. Dada la edad de los pacientes y la baja mortalidad, el control sin tratamiento puede ser una opción. En caso de linfedema es aconsejable la compresión elástica10.
- •
Radioterapia local. La radioterapia de bajo voltaje (100kV: 8Gy una sola aplicación o 20-30Gy dosis fraccionadas) es efectiva para las lesiones aisladas10. En un análisis retrospectivo de 68 pacientes tratados con radioterapia se observó una buena respuesta en el 85% de los pacientes, con respuesta completa en el 58% y una mejora de los síntomas en el 95%3.
- •
Extirpación quirúrgica. En caso de lesiones clínicamente molestas, como las de zonas acras, se aconseja la cirugía. En un estudio de pacientes tratados quirúrgicamente se comprobó que no se producían recidivas durante una media de 15meses tras el tratamiento3.
- •
Crioterapia. La crioterapia puede emplearse en lesiones únicas de tamaño ≤1cm, sobre todo de zonas acrales, con aplicaciones de 30-60s. Tanto la cirugía como la crioterapia tienen la ventaja de que pueden repetirse con buenos resultados.
- •
Terapia intralesional. El tratamiento intralesional de vinblastina (0,2mg/ml cada 2semanas), vincristina (0,03-0,08mg) o interferón alfa (3-5MUI/3 veces a la semana × 4-5semanas)10, la aplicación tópica de alitretinoína 0,1% gel (lesiones maculosas) o imiquimod 5% crema (3 veces por semana durante 24semanas)10 son tratamientos referidos en la literatura, aunque con escasa experiencia5.
- 2.
Enfermedad diseminada
- •
Doxorubicina pegilada liposomal (DPL) (20mg/m2 cada 3semanas). La DPL es el fármaco de elección, salvo cardiopatía. La respuesta es buena o excelente, con respuestas parciales (reducción del número de lesiones tumorales en un 50%) o completas durante 25meses en más del 70% de los pacientes11. La duración de la quimioterapia no está bien establecida, aunque se aconseja mantenerla 1-2ciclos después de obtener la respuesta clínica. El tratamiento con DPL es generalmente bien tolerado, con escasos efectos adversos, siendo menos cardiotóxico que el tratamiento con los compuestos tradicionales, lo que permite dosis acumulativas más altas y tratamientos más prolongados. Las toxicidades más graves (grado3 y 4) son infrecuentes e incluyen la neutropenia y la anemia12.
- •
Vinblastina (3mg/m2/semanal/i.v. o 6mg/m2/2semanas/i.v.). La vinblastina ofrece buenos resultados, con respuestas entre el 50 y el 90%, aunque puede producir leucopenia3,13.
- •
Otros quimioterápicos empleados con respuestas elevadas, pero con efectos adversos son: paclitaxel i.v. (100mg/semana), bleomicina (15U/semana ×3semanas y después cada 3semanas/i.m.) y etopósido oral (100mg/día, 3-5días a la semana)10. Solo existe un ensayo clínico aleatorizado entre vinblastina i.v. y etopósido oral en el que no se encontraron diferencias significativas en la respuesta o en la supervivencia, pero sí una mayor proporción de efectos adversos en el tratamiento con etopósido13.
Esquema de tratamiento del sarcoma de Kaposi
| Variante | Tratamiento | ||
|---|---|---|---|
| SK clásico | Tratamiento de elección | Otras alternativas | |
| I. Lesiones únicas o aisladas | a. Considerar solo observación clínica b. Radioterapia local o extirpación quirúrgica | Terapia intralesional | |
| II. Enfermedad diseminada | Doxorubicina liposomal | Vinblastina, placlitaxel, etopósido | |
| SK asociado a VIH | I. Lesiones únicas o aisladas | Iniciar TARGA con o sin terapia local (radioterapia, extirpación quirúrgica o crioterapia) | TARGA y terapia intralesional |
| II. Enfermedad diseminada | TARGA con doxorubicina liposomal | TARGA con paclitaxel | |
| SK iatrogénico (asociado a inmunosupresión) | Suspender o disminuir dosis de fármaco inmunosupresor | Seguir pauta de tratamiento de SK asociado a HIV | |
SK: sarcoma de Kaposi; TARGA: terapia antirretroviral de gran actividad.
- 1.
Lesiones únicas o limitadas
- •
TARGA en monoterapia o asociado a terapia local (fig. 4). El tratamiento inicial es la TARGA, ya que se ha demostrado que la reconstitución inmunológica reduce la incidencia y la gravedad del sarcoma2,7, con reducción o desaparición de las lesiones. En lesiones sintomáticas o antiestéticas se puede realizar cirugía14, electrocoagulación o crioterapia, y también aplicar vinblastina intralesional15 (0,2 a 0,3mg/ml, administrando 0,1ml por cada 0,5cm2 de lesión) o radioterapia de bajo voltaje (100kV, dosis entre 8Gy/1fracción o 30Gy/10fracciones, más del 95% de respuesta clínica completa)16. La supervivencia a los 5años del tratamiento con TARGA, con o sin terapia local, fue del 92% en una serie de más de 400 casos14.
- 2.
Enfermedad diseminada

Se recomienda tratamiento sistémico en aquellos pacientes tratados con TARGA y afectación cutánea extensa (más de 15-25 lesiones), edema intenso, SK cutáneo que no haya respondido a la terapia local o rápidamente evolutivo, SK asociado a síndrome de reconstitución inmunológica o afectación visceral sintomática.
- •
DPL (20mg/m2 cada 3 semanas). Debe iniciarse TARGA y DPL17, ya que la combinación es más eficaz que la TARGA sola18. Se tiende a realizar varias tandas de tratamiento según respuesta clínica. Se obtiene una respuesta completa/parcial con la terapia combinada en el 80%19, con supervivencias a los 5años de más del 85%. Las recaídas son escasas (13%) y se producen en el primer año tras la suspensión del fármaco20. La respuesta a DPL es superior a la combinación de bleomicina, vincristina o vinblastina y doxorubicina no liposomal21 y a la daunorubicina liposomal22.
- •
Paclitaxel (100mg/m2 cada 2 semanas). Es un fármaco de segunda línea con el que se obtienen respuestas favorables en el 71% de los pacientes23, pero con tasas de supervivencia inferiores a DPL y mayor toxicidad grados 3-514,24. Requiere premedicación con dexametasona y puede producir graves interacciones farmacológicas con los antirretrovirales.
- •
Otras terapias. Otros fármacos, como etopósido, vinorelbina, interleucina12, bevacizumab e imatinib también han sido empleados, pero la experiencia es limitada5.
- •
Suspender el fármaco inmunosupresor o disminuir su dosis. La suspensión o disminución del fármaco inmunosupresor puede inducir remisiones espontáneas. Si no fuera efectivo o fuera impracticable, generalmente se siguen las pautas utilizadas en el SK-VIH+5.
El SK es un tumor angioproliferativo con distintos subtipos asociados a edad avanzada, determinadas poblaciones africanas, inmunosupresión iatrogénica o VIH. Pese a que la TARGA ha producido un descenso dramático en su incidencia y gravedad en individuos VIH+, es importante conocer las distintas opciones terapéuticas según la variante de SK y su forma de presentación clínica.
Angiosarcoma cutáneoLos angiosarcomas representan entre el 1 y el 2%25,26 de todos los sarcomas, pero al menos la mitad de ellos son cutáneos. De los sarcomas cutáneos, el angiosarcoma es el cuarto en frecuencia, por detrás del sarcoma de Kaposi, el dermatofibrosarcoma y el leiomiosarcoma. El angiosarcoma cutáneo es una de las neoplasias cutáneas de peor pronóstico, con supervivencia a los 5años de entre el 10% en las series más antiguas27 y el 30 y el 50% en las más modernas25,28,29. Existen 3 grandes variedades de angiosarcomas cutáneos: los idiopáticos de cara y cuero cabelludo de pacientes ancianos (angiosarcoma de Wilson Jones), que suponen aproximadamente el 50% de los angiosarcomas cutáneos, y dos formas de angiosarcomas secundarios: una que asienta sobre áreas de linfedema crónico, especialmente en los brazos de mujeres sometidas a mastectomías radicales (síndrome de Stewart-Treves), y otra que se desarrolla sobre áreas de piel irradiada, especialmente en la región pectoral de mujeres sometidas a radioterapia tras cáncer de mama (fig. 5). Se trata de un sarcoma cutáneo muy agresivo con gran tendencia a la recidiva local y mal pronóstico27,30. El único tratamiento potencialmente curativo es la cirugía ±radioterapia.

La incidencia del angiosarcoma cutáneo es difícil de calcular, pero la de los angiosarcomas en su conjunto es de 0,4 casos por millón de habitantes en Estados Unidos31. Los angiosarcomas cutáneos suponen entre un 35 y un 60% de todos los angiosarcomas, con una incidencia aproximada de 0,2 casos por millón de habitantes. Predomina en pacientes ancianos con una media de edad de 73años, y es excepcional en niños o en pacientes jóvenes. El angiosarcoma clásico de Wilson Jones predomina en varones y el posradioterapia en mujeres29. Predomina en la raza blanca25,29. La mayoría de los angiosarcomas idiopáticos asientan en cabeza y cuello (62%), los posradioterapia predominan en el tronco (24%), especialmente en la zona pectoral (posradioterapia de la mama) y los poslinfedema en extremidades (11%). La mayoría de los angiosarcomas posradioterapia aparecen tras radioterapia por cáncer de mama32, pero hay casos en otras zonas radiadas y no solo por procesos malignos. El tiempo de latencia entre la radioterapia y el desarrollo del angiosarcoma es variable, con una media de 5años para la mama y de 10años o más para otras localizaciones. Los angiosarcomas poslinfedema predominan sobre linfedema crónico en los brazos de mujeres sometidas a mastectomías radicales33 (síndrome de Stewart-Treves), pero hay casos descritos sobre linfedemas de cualquier etiología. El tiempo transcurrido entre que se establece el linfedema y el desarrollo del angiosarcoma es muy variable: entre uno y 30años.
La forma inicial más característica es a modo de una lesión contusiforme, a veces edematosa, con frecuencia mal definida, que tiende a pasar desapercibida inicialmente, en especial los de cuero cabelludo de pacientes con pelo34. En los angiosarcomas de cabeza y cuello se aprecia mejor la extensión real del angiosarcoma si el paciente coloca su cabeza por debajo del nivel del corazón durante unos segundos. Así la porción subclínica se hace más visible al adquirir una tonalidad violácea y un aspecto edematoso35. Más adelante aparecen pápulas y nódulos, y ocasionalmente ulceración y sangrado en las fases avanzadas. Existen casos que se inician directamente con pápulas y nódulos sin apenas fase contusiforme previa. El tamaño medio al diagnóstico es de 3-5cm28,29. Los angiosarcomas pueden aparecer como un tumor único o multifocal y característicamente la extensión real sobrepasa los límites clínicamente apreciables. La sospecha clínica de angiosarcoma cutáneo ha de ser confirmada mediante biopsia.
Diagnóstico histológicoLos tres tipos de angiosarcomas cutáneos muestran características histopatológicas superponibles. Los angiosarcomas bien diferenciados muestran luces vasculares revestidas de endotelios aplanados que disecan entre los haces de colágeno, con poca atipia celular. En estas fases el diagnóstico histológico es complejo, y es útil reconocer algunos endotelios con células prominentes, pleomórficas, de núcleos hipercromáticos que tienden a protruir y a crear algunas papilas, con varias capas de células endoteliales en las luces vasculares36,37. Los canales vasculares son irregulares y tienden a anastomosarse entre sí (fig. 6). En los casos peor diferenciados las células tumorales son epitelioides o fusiformes, con atipias marcadas y abundantes mitosis y un patrón de crecimiento más sólido con pocas luces vasculares, de modo que se pueden confundir con carcinomas o incluso melanomas o fibrosarcomas. La presencia de vacuolas intracitoplasmáticas como expresión de una diferenciación vascular primitiva puede ser muy útil para sospechar el diagnóstico correcto en estos casos. Los angiosarcomas indiferenciados infiltran de forma muy destructiva y desaparecen los componentes normales de la dermis y los anejos cutáneos. Es frecuente un infiltrado inflamatorio acompañante parcheado, a veces tan denso que puede simular un linfoma38. La cantidad de hematíes y hemosiderina acompañante es muy variable. Es habitual encontrar distintos grados de diferenciación dentro de un mismo angiosarcoma. La epidermis puede ser normal, atrófica o estar ulcerada. Clásicamente el grado de diferenciación de los angiosarcomas cutáneos no se ha relacionado con el pronóstico, y es por ello que —a diferencia de otros sarcomas— el grado histológico no se tiene en cuenta para su estadificación39. Quizás esto sea revisado en el futuro, ya que en un trabajo reciente25 con la serie más larga de angiosarcomas cutáneos y de partes blandas publicado —con 821 pacientes—, desarrollan un modelo pronóstico que se correlaciona con la supervivencia y que incluye el grado histopatológico.
 La inmunohistoquímica corresponde a un angiosarcoma cutáneo que muestra positividad para ERG (típicamente de patrón nuclear). b) Angiosarcoma con predominio de áreas con patrón vasoformativo. c,d) En las fotos de detalle se muestran los endotelios neoplásicos, que en este caso resultan prominentes pero sin atipia llamativa.")
a) La inmunohistoquímica corresponde a un angiosarcoma cutáneo que muestra positividad para ERG (típicamente de patrón nuclear). b) Angiosarcoma con predominio de áreas con patrón vasoformativo. c,d) En las fotos de detalle se muestran los endotelios neoplásicos, que en este caso resultan prominentes pero sin atipia llamativa.
El estudio de un angiosarcoma ha de completarse con un panel inmunohistoquímico que incluye un panel básico de tumores fusocelulares (CD31, pancitoqueratinas, S100 y actina) y marcadores vasculares adicionales (CD34, ERG, podoplanina). Algunos casos de angiosarcomas con predominio de células epitelioides pueden mostrar alguna positividad para citoqueratinas; sin embargo, la positividad de marcadores vasculares como el CD31, el ERG y/o la podoplanina permitirá descartar un carcinoma indiferenciado. En los últimos años se ha demostrado que muchos angiosarcomas tienen amplificación/sobreexpresión del MYC. En las mayoría de los trabajos encuentran amplificación del MYC en torno al 50-100% de los angiosarcomas cutáneos secundarios y no en los idiopáticos40-42, pero también se ha demostrado amplificación o sobreexpresión del MYC en algunos angiosarcomas idiopáticos43,44. Sin embargo, prácticamente todos los estudios han demostrado la ausencia de amplificación o sobreexpresión del MYC en las proliferaciones vasculares atípicas posradioterapia, de modo que su positividad en un caso dudoso de proliferación vascular en área irradiada prácticamente descarta angiosarcoma.
El origen del angiosarcoma en vasos de naturaleza sanguínea o linfática es un tema controvertido. La expresión de CD31 o CD34 es mayor en las áreas mejor diferenciadas, pero ninguno de los dos es constante45. Los marcadores inmunohistoquímicos relativamente específicos de vasos linfáticos, como son la podoplanina, el D2-40, el LYVE-1 y el PROX-1, suelen ser positivos en los angiosarcomas cutáneos, de manera que pueden expresar en muchos casos un patrón inmunohistoquímico mixto tanto de endotelios sanguíneos como de endotelios linfáticos38,46,47. Aunque es excepcional, se han descrito casos de angiosarcomas cutáneos que expresan proteína S-10048 o marcadores neuroendocrinos49.
EstadificaciónAunque no existen guías de manejo del angiosarcoma cutáneo, dado que la localización más habitual de las metástasis es el pulmón50, seguida de los ganglios linfáticos29,51, se suele recomendar tras el diagnóstico clinicopatológico completar el estudio con una TAC toracoabdominal, e incluir la región cervical si se trata de un angiosarcoma de cabeza o cuello y la pelvis en angiosarcomas posradioterapia abdominopélvica. La presencia de diseminación regional o a distancia no es rara en el angiosarcoma y aparece en torno al 30 y al 10% de los casos, respectivamente29.
No existe un TNM específico para los angiosarcomas, de modo que se emplea el TNM de los sarcomas de partes blandas de la clasificación de la AJCC adaptado a los angiosarcomas. En el caso de los angiosarcomas, como actualmente no se acepta la influencia del grado histológico en el pronóstico, el estadioIA yIIA se agrupan en algunos trabajos lo mismo que los estadiosIB yIIB, que en otros sarcomas se distinguen entre sí solo por el grado histológico.
TratamientoEl único tratamiento que ha demostrado ser potencialmente curativo en el angiosarcoma cutáneo de forma aislada es la cirugía. No obstante, en casos inoperables o metastásicos la radioterapia y/o la quimioterapia tienen un papel paliativo reconocido. Además, en algunos trabajos recientes se incluye la radioterapia como adyuvante a la cirugía también en casos localizados de angiosarcomas50,51, e incluso algunos autores recomiendan irradiar los linfáticos regionales52, pero esto no se realiza habitualmente. El problema de la cirugía en el angiosarcoma cutáneo es que su carácter multicéntrico unas veces y su mala delimitación clínica otras, sumados al hecho frecuente de ser diagnosticados cuando su tamaño ya supera los 5cm, todo ello en el contexto de un paciente de avanzada edad, dificulta la obtención de unos márgenes quirúrgicos adecuados. Como norma general, si las características del tumor y el estado general del paciente lo permiten, el tratamiento del angiosarcoma cutáneo será la extirpación quirúrgica con márgenes adecuados. Lo más aceptado es la cirugía con 3cm de margen con respecto a los límites clínicamente apreciables53. El margen profundo no está bien establecido, dado que es un sarcoma dérmico, y parece razonable alcanzar hasta la fascia sin incluirla, pero en casos más infiltrativos deberá incluirse el músculo para alcanzar unos márgenes libres. Obtener márgenes afectos en el angiosarcoma es un factor de mal pronóstico demostrado en varios trabajos28,30,51. En el caso de la mama, la mayoría de los trabajos incluyen una mastectomía total a extirpaciones más o menos extensas de la piel irradiada. En casos complejos, un mapeo biópsico previo nos permitirá hacer un plan previo a la cirugía. Siempre que sea factible, en oncología cutánea se preferirán los cierres directos, los injertos o los cierres por segunda intención para facilitar el seguimiento y no enmascarar con plastias posibles recaídas locales, pero esto puede ser muy difícil o imposible tras extirpaciones radicales de angiosarcomas mamarios que impliquen mastectomía total y de toda la piel irradiada. Con respecto a los angiosarcomas poslinfedema, un artículo que revisa 160 pacientes publicados de síndrome de Stewart-Treves describe ausencia de beneficio de la amputación frente a la cirugía radical (con 2 o 3cm de margen) en estos casos, de modo que la amputación de la extremidad no parece justificada en angiosarcomas poslinfedema54.
En los casos en que la cirugía sea imposible —casos multicéntricos o muy extensos, o que afectan áreas de muy complejo abordaje quirúrgico—, la radioterapia será el tratamiento de elección55. La dosis de radioterapia para el angiosarcoma cutáneo es habitualmente de 60Gy repartidos en 20sesiones de 3Gy cada una. Cuando se usa de modo adyuvante a la cirugía, las dosis son similares, salvo en el caso de que la indicación sea para angiosarcomas posradioterapia, en los que habitualmente la dosis empleada será menor (en torno a 45-50Gy).
Con respecto a la quimioterapia, esta solo tiene un papel paliativo en el angiosarcoma, y se reserva para casos recidivados o metastásicos no susceptibles de cirugía. También se ha propuesto recientemente un papel neoadyuvante de la quimioterapia previa a la cirugía en localizaciones periorbitarias56. Los quimioterápicos que más se utilizan en el angiosarcoma son el docetaxel57,58, el paclitaxel59 y la doxorubicina liposomal60, pero se aceptan también en la guía actual de la NCCN la vinorelbina, el sorafenib, el sunitinib y el bevacizumab, pese a que los resultados con estos tres últimos antiangiogénicos son decepcionantes. La asociación de betabloqueantes en esta fase de tratamiento paliativo puede tener también algún beneficio para el paciente61,62.
SeguimientoNo existen normas estandarizadas en el seguimiento en los angiosarcomas cutáneos. Nosotros realizamos un seguimiento clínico más estrecho, con revisiones cada 3-6meses durante los tres primeros años, y posteriormente revisiones anuales durante 10años. En las visitas se explora toda la piel y se palpan los territorios ganglionares de drenaje correspondientes, y se realiza al menos una vez al año una analítica y una TAC toracoabdominopélvica.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



