






Kaposi sarcoma is a vascular sarcoma with 4 clinical variants: classic Kaposi sarcoma, which mainly affect the extremities of elderly patients and follows a chronic, generally indolent course; African Kaposi sarcoma; immunosuppression-associated Kaposi sarcoma; and AIDS-associated Kaposi sarcoma. Type8 human herpesvirus is the etiologic agent in all 4variants. Cutaneous angiosarcoma is a cutaneous neoplasm with a very poor prognosis. It carries a high probability of local relapse and has a 10% to 15% survival rate at 5years. There are 3 main variants of cutaneous angiosarcoma: idiopathic angiosarcoma of the face and scalp; Stewart-Treves syndrome; and postradiation angiosarcoma. The only potentially curative treatment is surgery with or without radiotherapy. However, its indistinct borders and multicentric nature mean that treatment is often palliative with chemotherapy, radiotherapy, or both.
El sarcoma de Kaposi es un sarcoma vascular con cuatro variantes clínicas: el clásico, que asienta preferentemente en las extremidades de pacientes ancianos, de curso crónico y poco agresivo; el endémico de África central; el de pacientes inmunodeprimidos, y el asociado a SIDA. En todas las variedades se ha demostrado que el virus herpes tipo8 es el agente etiológico. El angiosarcoma cutáneo es una de las neoplasias cutáneas de peor pronóstico, con gran tendencia a la recidiva local y una supervivencia a 5años del 10-50%. Existen 3 grandes variedades de angiosarcomas cutáneos: los idiopáticos de cara y cuero cabelludo, los desarrollados sobre áreas de linfedema crónico y los que aparecen sobre áreas de piel irradiada. El único tratamiento potencialmente curativo es la cirugía asociada o no a radioterapia, pero su mala delimitación y su carácter multicéntrico obligan en muchos casos a emplear tratamientos paliativos con quimio y/o radioterapia.
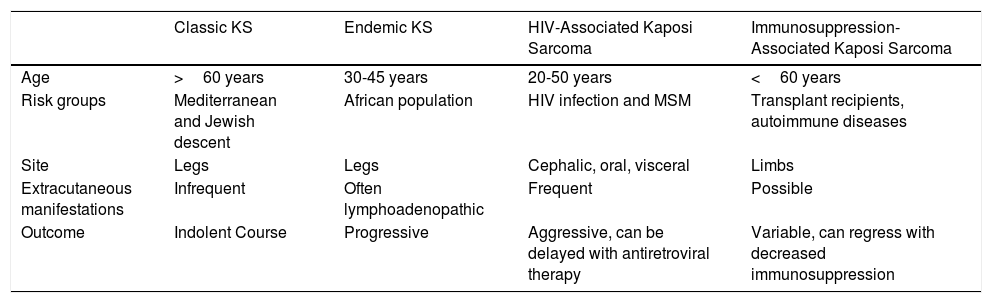
Kaposi sarcoma (KS) is an angioproliferative tumor associated with human herpesvirus type 8 virus (HHV-8).1,2 Four variants have been described (Table 1).
Epidemiology and Clinical Characteristics of the Different Types of Kaposi Sarcoma.
| Classic KS | Endemic KS | HIV-Associated Kaposi Sarcoma | Immunosuppression-Associated Kaposi Sarcoma | |
|---|---|---|---|---|
| Age | >60 years | 30-45 years | 20-50 years | <60 years |
| Risk groups | Mediterranean and Jewish descent | African population | HIV infection and MSM | Transplant recipients, autoimmune diseases |
| Site | Legs | Legs | Cephalic, oral, visceral | Limbs |
| Extracutaneous manifestations | Infrequent | Often lymphoadenopathic | Frequent | Possible |
| Outcome | Indolent Course | Progressive | Aggressive, can be delayed with antiretroviral therapy | Variable, can regress with decreased immunosuppression |
Abbreviations: MSM, men who have sex with men; KS, Kaposi sarcoma.
Classic Kaposi sarcoma. This is an uncommon tumor that tends to affect men1,3 in the Mediterranean or central European region, with an incidence of between 0.18 and 13.2 cases/million.4 It occurs more frequently in men with chronic leg edema, diabetes mellitus, and corticosteroid users. The lesions present as single or multiple slow-growing erythematous-violaceous plaques or nodules, on rare occasions associated with lymphedema in the extremities and gastrointestinal and lymph node involvement. It has an indolent clinical course, and 2% of the patients die of disseminated disease.
Endemic Kaposi sarcoma. This variant has been reported in equatorial Africa and affects young adults and prepubescent individuals. In adults, the variant follows an indolent or aggressive course, with involvement of subcutaneous and bone tissue, whereas in children it is manifest in an aggressive form with generalized lymph node involvement, involvement of internal organs, and absence of (or limited) skin lesions.5
Immunosuppression-associated Kaposi sarcoma (iatrogenic variant). This variant has been reported in patients in treatment with immunosuppressants and in transplant recipients in particular. The risk of developing KS is estimated to be between 150 and 200 times greater than in the general population, with a mean time to onset of 18months.6
HIV-associated Kaposi sarcoma (epidemic variant). This variant has been reported in men with HIV infection who have sex with men (MSM-HIV+). Before the era of highly active antiretroviral therapy (HAART), it was calculated that 25% of MSM-HIV+ would develop KS, although this percentage has decreased progressively2,7 (Figs. 1 and 2). These patients may experience involvement of skin and mucosa, lymph nodes, gastrointestinal tract, lungs, spleen, and liver.7 KS in MSM-HIV- has also been reported, in this case with an indolent course.8


Diagnosis is performed clinically, but it is recommended to obtain confirmation by biopsy. Histological study reveals spindle cells, proliferation of irregular vessels with slit-like forms, red blood cell extravasation, and leukocytic infiltrate with plasma cells and intra- and extracellular hyalin globules throughout the thickness of the dermis, as well as the so-called promontory sign (Fig. 3). Polymerase chain reaction and immunohistochemical staining for latency-associated nuclear antigen (LANA-1) of the HHV-8 virus are positive.1

Histology of Kaposi sarcoma.
A, Low-magnification view of Kaposi sarcoma in nodular phase. Well-defined dermal nodule.
B, Highly cellular lesion with some gaps in the form of cracks.
C, Detail of spindle cells and red blood cells within the small vessels.
D, Immunohistochemical nuclear positivity for HHV-8 specific to Kaposi sarcoma.
Classic Kaposi sarcoma. In view of the clinical presentation (age, local involvement, infrequent involvement of internal organs, and indolent course), skin and lymph node examination is sufficient. Complementary tests are performed if the patient presents symptoms of visceral compromise.5
Immunosuppression-associated Kaposi sarcoma. As for HIV-associated KS discussed below, there is no consensus staging for immunosuppression-associated KS. Given the lack of consensus, the tests and recommended applicable criteria are usually the same as for HIV-associated KS.
HIV-Associated Kaposi Sarcoma There is no accepted staging system for this variant. Chest X-ray is recommended and, in the event of abnormalities suggestive of respiratory compromise, bronchoscopy or computed chest tomography should be performed. It is also recommended to rule out fecal occult blood; if this test is positive, digestive endoscopy should be performed.
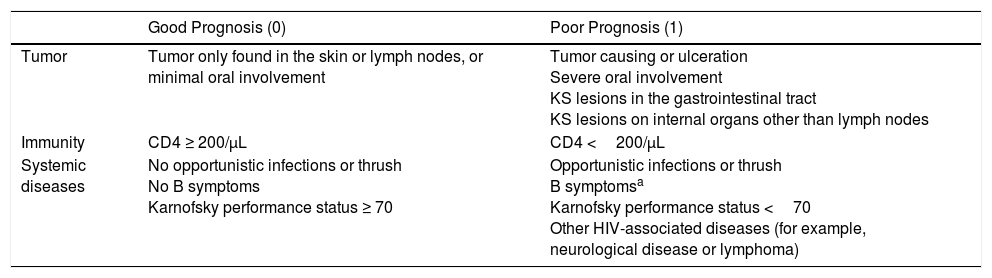
In 1989, the AIDS Clinical Trials Group Oncology Committee proposed staging (Table 2) based on disease extension, CD4T-cell count, and systemic compromise (opportunistic infections, B symptoms such as fever, weight loss, or persistent diarrhea, and Karnofsky performance status below 70 points). A prospective analysis showed that these variables were related to patient survival, with factors associated with good prognosis being limited disease, CD4 lymphocyte count greater than 150cells/mm3, and absence of systemic compromise.9
Staging Proposed by the AIDS Clinical Trials Groups Oncology Committee.
| Good Prognosis (0) | Poor Prognosis (1) | |
|---|---|---|
| Tumor | Tumor only found in the skin or lymph nodes, or minimal oral involvement | Tumor causing or ulceration Severe oral involvement KS lesions in the gastrointestinal tract KS lesions on internal organs other than lymph nodes |
| Immunity | CD4 ≥ 200/μL | CD4 <200/μL |
| Systemic diseases | No opportunistic infections or thrush No B symptoms Karnofsky performance status ≥ 70 | Opportunistic infections or thrush B symptomsa Karnofsky performance status <70 Other HIV-associated diseases (for example, neurological disease or lymphoma) |
Abbreviation; KS, Kaposi sarcoma.
These proposals for staging were put forward before HAART was available and do not include viral load, and so application of this staging system has been limited to clinical trials.
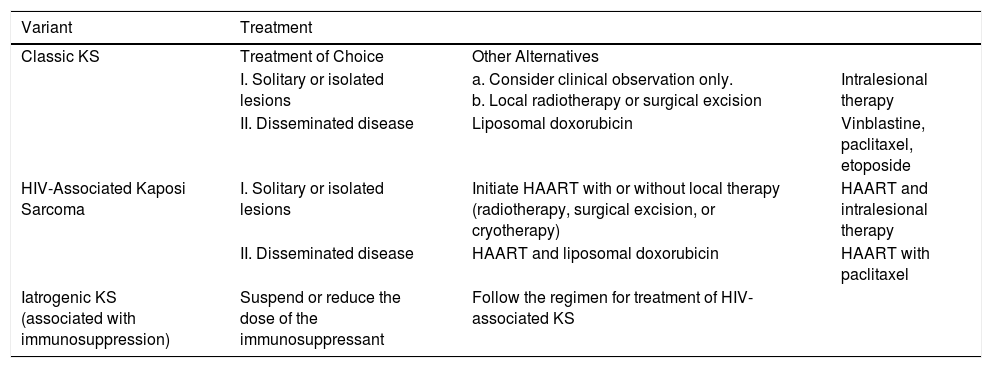
TreatmentClassic Kaposi SarcomaThere are few comparative clinical trials of the different treatments for classic KS. The same drugs and regimens as those applied for epidemic KS are usually used (Table 3).
- 1.
Solitary lesions.
- •
Clinical observation. Given the age of the patients and low mortality, follow-up without treatment can be an option. In the case of lymphedema, elastic compression is recommended.10
- •
Local radiotherapy. Low-energy radiotherapy (100kV: 8Gy in a single application or 20-30Gy as a fractionated dose) is effective for isolated lesions.10 In a retrospective analysis of 68 patients treated with radiotherapy, good response was observed in 85% of patients, with complete response in 58% and improvement in symptoms in 95%.3
- •
Surgical excision. In the case of lesions that cause discomfort, such as those in acral areas, surgery is recommended. In a study of surgically excised lesions, patients were found to be relapse-free for a mean of 15 months after treatment.3
- •
Cryotherapy. Cryotherapy can be used in solitary lesions measuring ≤1cm, particularly in acral regions, with applications lasting from 30 to 60 seconds. Both surgery and cryotherapy have the advantage that they can be repeated with good outcomes.
- •
Intralesional therapy. Intralesional treatment with vinblastine (0.2mg/mL every 2weeks), vincristine (0.03-0.08mg), or interferon alfa (3-5MIU/3 times a week for 4-5weeks),10 topical application of alitretinoin 0.1% gel (macular lesions) or imiquimod 5% cream (3 times a week for 24weeks)10 are treatments recommended in the literature, although there is limited experience with these regimens.5
- 2.
Disseminated Disease
- •
Pegylated liposomal doxorubicin (PLD) (20mg/m2 every 3weeks). PLD is the treatment of choice except in patients with heart disease. Response is good or excellent, with partial responses (50% decrease in number of tumor lesions) or complete response lasting 25 months in more than 70% of patients.11 The duration of chemotherapy is not well established, although it is recommended to maintain treatment for 1-2 cycles after clinical response. Treatment with PLD is generally well tolerated, with limited side effects, and it is less cardiotoxic than treatment with traditional compounds. Higher cumulative doses and longer treatments can therefore be administered. The most severe toxicities (grade 3 and 4) are infrequent and include neutropenia and anemia.12
- •
Vinblastine (3mg/m2/week/intravenously [iv] or 6mg/m2/2weekly/iv). Vinblastine offers good outcomes, with responses rates between 50% and 90%, although leukopenia may occur.3,13
- •
Other chemotherapies associated with high response rates, but with adverse effects include paclitaxel iv (100mg/week), bleomycin (15U/week for3weeks and then every 3weeks/intramuscularly), and oral etoposide (100mg/day, 3-5days a week).10 There is only 1 randomized clinical trial that has compared vinblastine iv with oral etoposide; no significant differences were found in response or survival, but a higher proportion of adverse effects were reported with etoposide treatment.13
Algorithm for Treatment of Kaposi sarcoma.
| Variant | Treatment | ||
|---|---|---|---|
| Classic KS | Treatment of Choice | Other Alternatives | |
| I. Solitary or isolated lesions | a. Consider clinical observation only. b. Local radiotherapy or surgical excision | Intralesional therapy | |
| II. Disseminated disease | Liposomal doxorubicin | Vinblastine, paclitaxel, etoposide | |
| HIV-Associated Kaposi Sarcoma | I. Solitary or isolated lesions | Initiate HAART with or without local therapy (radiotherapy, surgical excision, or cryotherapy) | HAART and intralesional therapy |
| II. Disseminated disease | HAART and liposomal doxorubicin | HAART with paclitaxel | |
| Iatrogenic KS (associated with immunosuppression) | Suspend or reduce the dose of the immunosuppressant | Follow the regimen for treatment of HIV-associated KS | |
Abbreviations: HAART, highly active antiretroviral therapy; KS, Kaposi sarcoma
- 1.
Solitary or Limited Lesions
- •
HAART in monotherapy or associated with local therapy (Fig. 4). HAART is administered as the initial treatment, given that such agents have been shown to reconstitute immune function and lower the incidence and severity of the sarcoma,2,7 with reduction or disappearance of lesions. In symptomatic and antiesthetic lesions, surgery,14 electrocoagulation, or cryotherapy are options. Intralesional vinblastin15 (0.2 to 0.3mg/mL, administering 0.1mL for every 0.5cm2 lesion) or low-energy radiotherapy (100kV, doses between 8Gy/1fraction or 30Gy/10fractions, more than 95% complete clinical response) can be considered.16 Lesion-free survival at 5 years after HAART treatment, with or without local therapy was 92% in a series of more than 400 cases.14
- 2.
Disseminated Disease

Systemic treatment is recommended in those patients treated with HAART and with extensive skin involvement (more than 15 to 25 lesions), intense swelling, cutaneous KS that has not responded to local therapy or that is progressing rapidly, KS associated with immune reconstitution syndrome, or symptomatic involvement of internal organs.
- •
PLD (20mg/m2 every 3weeks). HAART and PLD should be started at the same time,17 as the combination is more effective than HAART alone.18 Several courses of treatment are usually administered depending on clinical response. Complete/partial response is obtained with combination therapy in 80%,19 with 5 year survival greater than 85%. Relapses are limited (13%) and occur in the first year after finishing treatment.20 PLD response is higher than the combination of bleomycin, vincristine, or vinblastine and nonliposomal doxorubicin,21 and liposomal daunorubicin.22
- •
Paclitaxel (100mg/m2 every 2 weeks). Paclitaxel is a second-line treatment with favorable responses in 71% of patients,23 but with survival rates lower than PLD and higher rates of grade 3-5 toxicities.14,24 Premedication with dexamethasone is required and severe drug-drug interactions may occur with antiretroviral agents.
- •
Other therapies. Other drugs, such as etoposide, vinorelbine, interleukin12, bevacizumab, and imatinib have also been used, but experience is limited.5
- •
Suspend the immunosuppressant or reduce the dose. Suspension or reduction of the immunosuppressant dose may induce spontaneous remissions. If this is ineffective or impractical, the approaches used in HIV-associated KS are usually applied.5
KS is an angioproliferative tumor with different subtypes associated with advanced age, certain African populations, iatrogenic immunosuppression, or HIV infection. Although HAART has led to a dramatic decrease in the incidence and severity of KS in individuals with HIV infection, it is important to be aware of the different therapeutic options according to the KS variant and its clinical presentation.
Cutaneous AngiosarcomaAngiosarcomas represent between 1% and 2% of all sarcomas, but at least half are cutaneous.25,26 Of the cutaneous sarcomas, angiosarcoma is the fourth most frequent, behind KS, dermatofibrosarcoma, and leiomyosarcoma. Cutaneous angiosarcoma is one of the cutaneous neoplasms with worst prognosis, with 5-year survival between 10% in the oldest series27 and 30%-50% in the most recent ones.25,28,29 There are 3 main variants of cutaneous angiosarcomas: idiopathic lesions on the face and scalp of elderly patients (Wilson-Jones angiosarcoma), a variant which accounts for approximately 50% of cutaneous angiosarcomas, and 2 forms of secondary angiosarcoma, one localized in areas of chronic lymphedema, particularly in the arms of women who undergo radical mastectomy (Stewart-Treves syndrome) and another that develops over areas of irradiated skin, particularly in the pectoral area of women who undergo radiotherapy after breast cancer (Fig. 5). This is a very aggressive cutaneous sarcoma with frequent local recurrence and poor prognosis.27,30 The only potentially curative therapy is surgery with or without radiotherapy.

The incidence of cutaneous angiosarcoma is difficult to calculate, but angiosarcomas together are reported at a rate of 0.4 cases per million inhabitants in the United States.31 Cutaneous angiosarcomas account for between 35% and 60% of all angiosarcomas, with an approximate incidence of 0.2 cases per million inhabitants. They predominate in elderly patients with a mean age of 73 years. These lesions are very rare in children or young patients. The classic Wilson-Jones angiosarcoma predominates in men and after radiotherapy in women.29 It also predominantly affects whites.25,29 The majority of idiopathic angiosarcomas are located on the head and neck (62%), postradiation lesions predominate on the trunk (24%), particularly in the pectoral area (postradiation of the breast) and after lymphedema on the limbs (11%). Most postradiation angiosarcomas appear after radiotherapy due to breast cancer,32 but there are cases in other irradiated areas and not just because of malignant processes. The latency time between radiotherapy and the development of angiosarcoma varies, with a mean of 5 years for breast sites and 10 years for other sites. Angiosarcomas due to lymphedema predominately occur in areas of chronic lymphedema on the arms of women who undergo radical mastectomy (Stewart-Treves syndrome),33 but cases have been reported of lesions over lymphedema of any etiology. The time between presentation of lymphedema and development of angiosarcomas varies greatly between 1 year and 30 years.
The initial most characteristic form is a bruise-like lesion, at times edematous, with a poor defined frequency, that tends to go unnoticed initially, particularly on the scalp of patients with hair.34 In the case of angiosarcoma of the head and neck, the true extent of the lesion can be better appreciated if the patient holds his or her head below the level of the heart for a few seconds. This maneuver will make the subclinical part become more visible as it takes on a violaceous tone and edematous appearance.35 Later in the clinical course, papules and nodules appear, occasionally with ulceration and bleeding in advanced phases. Some cases present directly with papules and nodules with hardly any prior bruise-like phase. The mean size on diagnosis is 3-5 cm.28,29 Angiosarcomas can appear as a solitary or multifocal tumor and the lesion will often extend beyond the clinically appreciable limits. Clinical suspicion of cutaneous angiosarcoma should be confirmed by biopsy.
Histologic DiagnosisThe 3 types of cutaneous angiosarcoma have overlapping histopathologic characteristics. Well-differentiated angiosarcomas show vascular lumina lined by flattened endothelial cells, dissecting collagen bundles, with limited cell atypia. Histologic diagnosis is complex in these phases and it is useful to recognize some endothelia with prominent, pleomorphic cells with hyperchromatic nuclei that tend to protrude and create some papillae, with several endothelial cell layers in the vascular lumina.36,37 The vascular conduits are irregular and tend to form anastomosing channels (Fig. 6). In the worst differentiated cases, tumor cells are epithelioid or fusiform, with marked atypia and abundant mitosis and a more solid growth pattern with few vascular lumina, such that they can be confused with carcinoma or even melanomas or fibrosarcomas. The presence of intracytoplasmic vacuoles as expression of primitive vascular differentiation can be very useful for suspicion of correct diagnosis in these cases. Infiltration in the case of undifferentiated angiosarcomas is very destructive and normal components of the dermis and skin appendages disappear. A patchy inflammatory infiltrate often accompanies the process and this infiltrate may at times be so dense that it resembles lymphoma.38 The extent of accompanying red blood cells and hemosiderin is very variable. Different degrees of differentiation may often be found in the same angiosarcoma. The epidermis may be normal, atrophic, or ulcerated. The extent of differentiation of cutaneous angiosarcomas is not traditionally thought to have an impact on prognosis, and so, unlike with other sarcomas, the histologic grade is not taken into account for staging.39 This approach may change in the future, as a recent study with the largest series of cutaneous angiosarcomas and soft tissue angiosarcomas published to date, with 821 patients, developed a prognostic model for survial which included histopathologic grade.25
. B, Angiosarcoma with predominance of areas with vasoformative pattern. C, D, Detailed images of neoplastic endothelial cells, which in this case are prominent but without noteworthy atypia.")
A, Immunohistochemistry of cutaneous angiosarcoma that is positive for ERG (typically with a nuclear pattern). B, Angiosarcoma with predominance of areas with vasoformative pattern. C, D, Detailed images of neoplastic endothelial cells, which in this case are prominent but without noteworthy atypia.
Study of angiosarcoma should be completed with an immunohistochemical panel that includes a basic panel for spindle-cell tumors (CD31, pancytokeratins, S110, and actin) and additional vascular markers (CD34, erythroblast transformation-specific–related gene [ERG], podoplanin). Some cases of angiosarcoma with predominance of epithelioid cells may be positive for cytokeratins; however, positivity for vascular markers such as CD31, ERG, and/or podoplanin can rule out undifferentiated carcinoma. In recent years, it has been shown that many angiosarcomas have MYC amplification/overexpression. In most studies, MYC amplification is found in between 50% and 100% of secondary angiosarcomas but not generally in idiopathic ones,40–42 but MYC amplification or overexpression has also been detected in some idiopathic angiosarcomas.43,44 However, almost all studies have shown the absence of MYC amplification or overexpression in atypical postradiation vascular proliferations, and so positivity in a doubtful case of vascular proliferation in the irradiated area almost rules out angiosarcoma.
The origin of angiosarcoma in blood vessels or lymphatic vessels is subject to debate. Expression of CD31 or CD34 is greater in the most differentiated areas, but in neither case is it constant.45 Immunohistochemical markers relatively specific for lymphatic vessels, such as podoplanin, D2-40, LYVE-1, and PROX-1, are usually positive in cutaneous angiosarcomas, often expressed with a mixed immunohistochemical pattern of endothelial blood vessels and endothelial lymphatic vessels.38,46,47 Although exceptional, cases have been reported of cutaneous angiosarcomas that express S-100 protein,48 or neuroendocrine markers.49
StagingAlthough there are no guidelines for the management of cutaneous angiosarcoma, given that the most frequent site of metastasis is the lung50 followed by the lymph nodes,29,51 thoracoabdominal computed tomography is usually recommended after pathological diagnosis. This imaging study should include the cervical region in the case of an angiosarcoma of the head and neck and the pelvis in in the case of abdominopelvic postradiation angiosarcomas. The presence of regional or distant dissemination is not uncommon with angiosarcomas, and is reported in 30% and 10% of cases, respectively.29
There is no TNM staging specific to angiosarcomas, and so the TNM staging for soft tissue sarcomas of the American Joint Committee on Cancer classification, adapted to angiosarcomas, is used. In the case of angiosarcomas, as histologic grade is not currently considered to impact prognosis, stage IA andIIA is grouped in some studies with stages IB andIIB, which in other sarcomas are differentiated only by histologic grade.
TreatmentThe only treatment shown to be potentially curative in cutaneous angiosarcoma is surgery, although cure only occurs sporadically. However, in inoperable or metastatic cases, radiotherapy and/or chemotherapy have a recognized palliative role. Moreover, some recent studies have also included radiotherapy as an adjuvant to surgery in localized cases of angiosarcoma,50,51 and some authors have even recommended to irradiate regional lymph nodes,52 but this is not common practice. The problem with surgery in cutaneous angiosarcoma is at times the multicenter character and the poor clinical delimitation in others, as well as the fact that they are often diagnosed when lesions exceed 5 cm in size. In addition to these factors, patients are often elderly which makes it more difficult to obtain suitable surgical margins. In general, if the tumor characteristics and general state of the patient permit, treatment of cutaneous angiosarcoma is surgical excision with sufficient margins. The most accepted is surgery with 3 cm margin with respect to the clinically appreciable limits.53 The depth of the margin is not well established, given that this is a dermal sarcoma, though it would appear reasonable to reach the fascia without excising them. In the most infiltrative cases, however, the muscle should be included to achieve clear margins. Margins with angiosarcoma involvement have been shown to be a factor for poor prognosis in several studies.28,30,51 In the case of breast involvement, most studies suggest total mastectomy or more or less extensive excisions of irradiated skin. In complex cases, prior biopsy mapping can help assist with presurgical planning. Whenever possible, in cutaneous oncology, direct closure, grafts, or second intent closures are preferred to facilitate follow-up and not mask possible local recurrence with surgical reconstruction, but this may be difficult or impossible after more radical excisions of mammary angiosarcomas that require total mastectomy with irradiation of the entire skin. In the case of angiosarcomas caused by lymphedema, a study reviewed reports of 160 patients with Stewart-Treves syndrome and found absence of benefit of amputation compared with radical surgery (with 2 or 3 cm margin) in these cases, and so limb amputation does not appear to be justified in such angiosarcomas.54
In cases in which surgery is impossible, that is multicentric or extensive lesions, or those that affect areas that complicate surgery, radiotherapy is the treatment of choice.55 The dose of radiotherapy for cutaneous angiosarcoma is usually 60 Gy distributed in 20 sessions of 3 Gy each. When used as an adjuvant to surgery, the doses are similar, except when indicated for postradiation angiosarcomas, in which the dose will usually be lower (45-50 Gy).
The only role of chemotherapy in angiosarcoma is as a palliative treatment, reserved for relapsed or metastatic lesions not amenable to surgery. A neoadjuvant role has also recently been proposed for chemotherapy prior to surgery in periorbital sites.56 The most widely used chemotherapies in angiosarcoma are docetaxel,57,58 paclitaxel,59 and liposomal doxorrubicin,60 but the current NCCN guidelines also include vinorelbine, sorafenib, sunitinib, and bevacizumab, although the results with these latter 3 antiangiogenic agents have been disappointing. Combination with beta blockers in this phase of palliative treatment may be of some benefit for the patient.61,62
Follow-upThere are no standard guidelines for follow-up of cutaneous angiosarcomas. In our group, we have close clinical follow-up, with check-ups every 3-6 months for the first few years, and then yearly check-ups for 10 years. In these visits, we examine the entire skin and palpate the corresponding territorial lymph nodes. At least once a year, we perform a laboratory analysis and thoracoabdominopelvic computed tomography study.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Requena C, Alsina M, Morgado-Carrasco D, Cruz J, Sanmartín O, Serra-Guillén C, et al. Sarcoma de Kaposi y angiosarcoma cutáneo: directrices para el diagnóstico y tratamiento. Actas Dermosifiliogr. 2018;109:878–887.



