



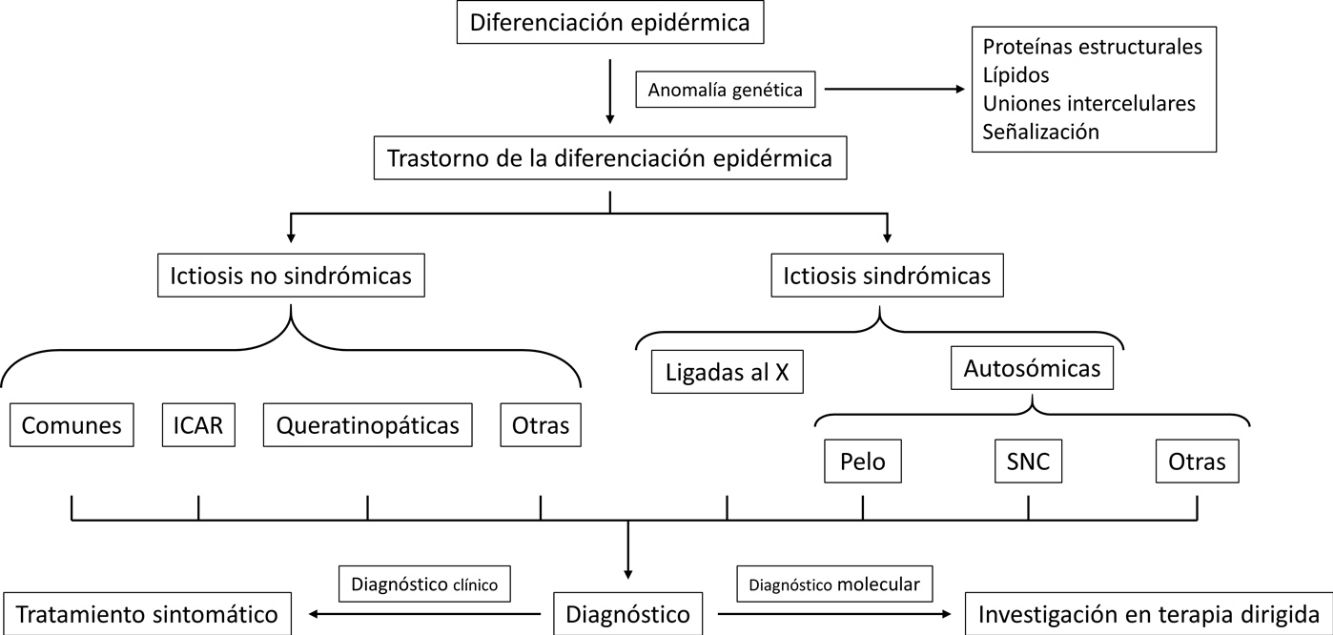



Las ictiosis son un grupo heterogéneo de enfermedades que comparten síntomas y un mismo mecanismo etiopatogénico. Desde el punto de vista clínico estas enfermedades se caracterizan por la presencia de eritema y diferentes grados de engrosamiento y descamación cutáneas. Aunque el área afectada, la gravedad y el sustrato molecular son muy variables, todas ellas representan la manifestación de una disrupción de la barrera que se forma durante el proceso de diferenciación epidérmica. Las ictiosis siguen patrones de herencia mendeliana y causan síntomas desde el nacimiento o poco tiempo después. Desde el punto de vista clínico se dividen en ictiosis no sindrómicas (cuando los síntomas están causados únicamente por el defecto de la barrera epidérmica) e ictiosis sindrómicas (cuando el gen causal también tiene funciones extracutáneas que determinan manifestaciones en otros órganos).
El conocimiento de las bases moleculares ha avanzado extraordinariamente en los últimos años, y conocemos no solo la mayoría de los genes que las ocasionan, sino la función de las proteínas que codifican y el impacto en la formación de la barrera cutánea. En la primera parte de este trabajo hacemos una introducción a la fisiopatología de las ictiosis, así como una actualización clínica y genética de las entidades no sindrómicas, tanto las incluidas en la última clasificación consenso como otras que se han caracterizado clínica y/o molecularmente a lo largo de los últimos años.
Ichthyoses are a heterogeneous group of diseases sharing symptoms and a common etiopathogenic mechanism. Clinically, these diseases are characterized by the presence of erythema and variable degrees of skin thickening and desquamation. Although the affected area, severity, and molecular substrate are very variable, they are all signs of a disruption of the barrier formed during epidermal differentiation. Ichthyoses follow patterns of Mendelian inheritance and present symptoms since birth or shortly thereafter. Clinically, they can be categorized into non-syndromic (when symptoms are caused exclusively by the epidermal barrier dysfunction) and syndromic ichthyoses (when the causal gene has extracutaneous functions impacting other organs).
Knowledge of molecular mechanisms has improved dramatically over the past few years, and we currently know not only most causal genes, but also the functions of the encoded proteins and their impact on skin barrier formation. In the first part of this review, we’ll be introducing ichthyosis physiopathology, along with a clinical and genetic update of non-syndromic entities (those included in the consensus classification and those clinically and/or molecularly characterized since then).

Las ictiosis son un grupo heterogéneo de enfermedades que comparten síntomas y un mismo mecanismo etiopatogénico. Desde el punto de vista clínico estas enfermedades se caracterizan por la presencia de eritema y diferentes grados de engrosamiento y descamación cutáneas. Aunque el área afectada, la gravedad y el sustrato molecular son muy variables, todas ellas representan la manifestación de una disrupción de la barrera que se forma durante el proceso de diferenciación epidérmica. Por tanto, este grupo de enfermedades se agrupan bajo el nombre de trastornos de la diferenciación epidérmica (TDE)1.
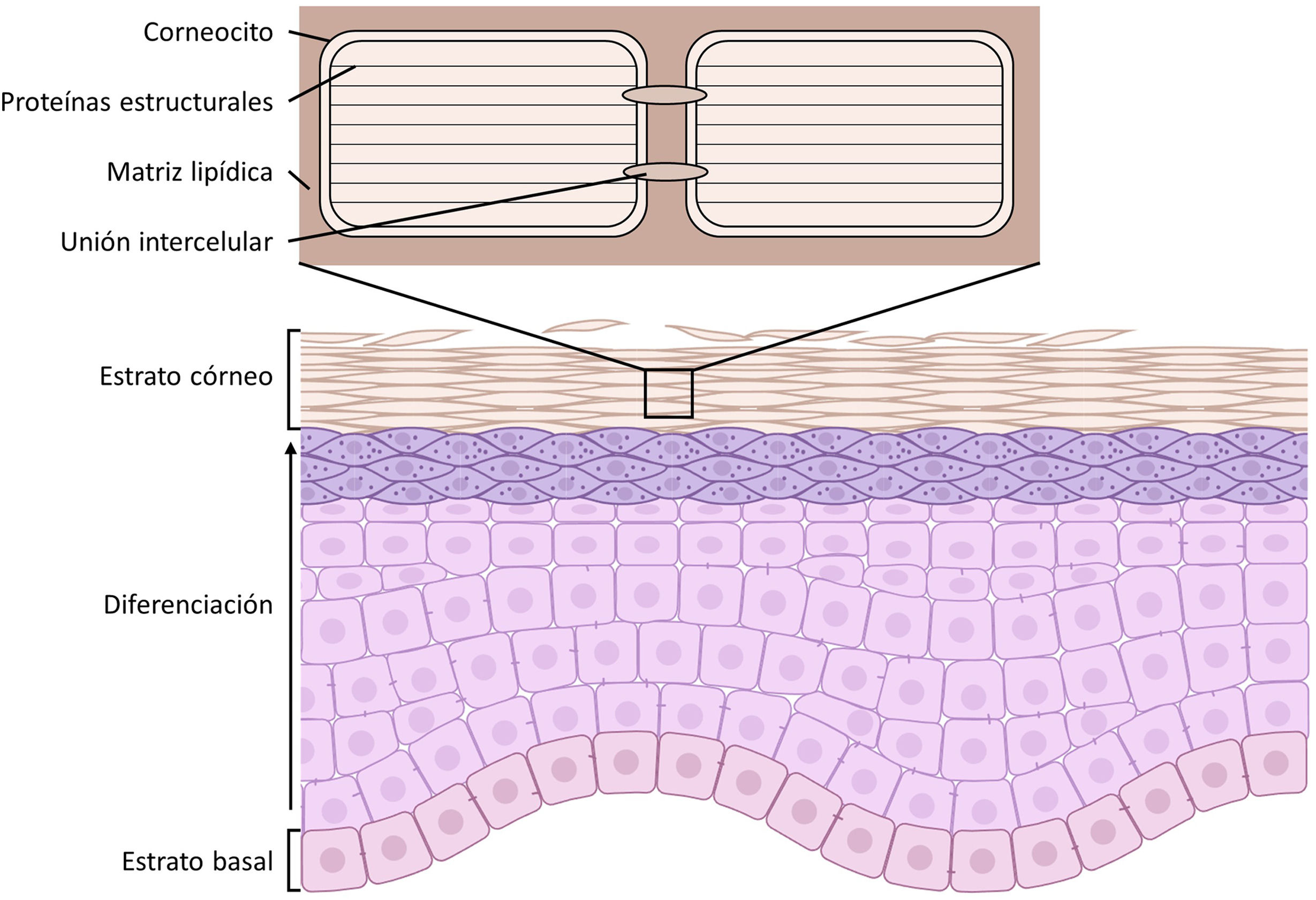
La epidermis es un tejido con una estructura muy bien definida: las células madre en constante división encargadas de regenerar el epitelio (queratinocitos) se localizan en el estrato basal y las células generadas a partir de esta división sufren un proceso gradual de diferenciación a medida que migran hacia la superficie2. Este proceso de diferenciación (denominado cornificación) causa cambios morfológicos en los queratinocitos debido a cambios en los patrones de expresión de proteínas, reorganización del citoesqueleto, secreción de lípidos y establecimiento de uniones intercelulares, culminando con la formación del estrato córneo. El estrato córneo es la capa más superficial de la epidermis, en la que los queratinocitos se han diferenciado terminalmente a corneocitos, células carentes de núcleo y orgánulos y cuyo citoplasma está completamente repleto de proteínas estructurales. Los corneocitos forman uniones entre ellos denominadas corneodesmosomas y se encuentran embebidos en una matriz lipídica secretada en las capas inferiores que satura el espacio intercelular y en la que se encuentran inmersas enzimas capaces de degradar las uniones intercelulares3 (fig. 1).

Estructura de la epidermis. Las células madre del estrato basal se diferencian hasta dar lugar al estrato córneo, compuesto de los corneocitos repletos de proteínas estructurales, anclados mediante uniones intercelulares y embebidos en una matriz lipídica extracelular. Fuente: modificado de «Ichthyosis», C. Gutiérrez-Cerrajero et al.9.
El proceso de cornificación genera una estructura orgánica cuya función es actuar de barrera entre el medio y el resto del organismo. Las proteínas estructurales confieren a las células resistencia mecánica frente a agresiones físicas y químicas; los corneodesmosomas unen las células y transfieren esta resistencia mecánica a todo el tejido; los lípidos sellan los espacios intercelulares, evitando el intercambio de agua y, por tanto, tanto la entrada de patógenos como la pérdida de agua por evaporación; y, finalmente las enzimas secretadas al espacio intercelular se encargan de la degradación ordenada de las uniones intercelulares, a fin de permitir la descamación del epitelio, lo que dificulta la colonización de la piel por parásitos. Además, el pequeño grosor del estrato córneo hace de la barrera epidérmica una estructura muy flexible, capaz de llevar a cabo sus funciones sin limitar el movimiento del organismo4.
Una alteración de la barrera epidérmica que impide que lleve a cabo sus funciones expone al organismo a agresiones físicas, químicas y biológicas y permite la evaporación transepidérmica de agua. Como respuesta homeostática, el organismo aumenta la producción de células diferenciadas y engrosa el estrato córneo. Esto lleva a la aparición del signo común de los TDE, la hiperqueratosis y el aumento de la descamación. El engrosamiento del estrato córneo causa pérdida de elasticidad, de la piel, lo que dificulta la movilidad y puede llevar a la formación de fisuras. La evaporación transepidérmica de agua conlleva riesgo de deshidratación y, junto con la hipohidrosis asociada a estos trastornos, intolerancia al calor. Además, el defecto en la barrera aumenta la exposición del sistema inmune a patógenos, lo que causa una hiperactivación de las rutas inflamatorias, eritema y prurito5,6.
Cuando esta alteración de la barrera epidérmica se debe a factores extrínsecos, la enfermedad se denomina ictiosis adquirida. Es un hallazgo asociado a determinados tumores (enfermedad de Hodgkin, mieloma múltiple y linfoma de células T cutáneo), enfermedades endocrinas (fracaso renal, diabetes, hiperparatiroidismo), enfermedades autoinmunes (lupus eritematoso y dermatomiositis), enfermedades infecciosas (lepra, infecciones por micobacterias o VIH), trastornos nutricionales (normalmente del metabolismo lipídico o de vitaminas) o ciertas medicaciones (reductores de colesterol, alopurinol, acitretina e inhibidores de EGFR y BRAF). La ictiosis adquirida es de aparición tardía (años o décadas después del nacimiento), sus síntomas suelen ser leves comparados con el del resto de los TDE, y su tratamiento suele dirigirse contra el factor extrínseco causante de la enfermedad7
Los TDE de origen genético, responden a patrones de herencia mendelianos y causan síntomas desde el nacimiento o pocos meses después. En la actualidad se han descrito más de cien genes cuyas variantes patogénicas causan algún tipo de TDE. La presentación fenotípica de estas enfermedades es muy heterogénea, ya que esta depende de las funciones de la proteína afectada. En la tabla 1 se expone la función en la diferenciación epidérmica de los genes involucrados en la aparición de las ictiosis1. En consecuencia, el diagnóstico clínico puede resultar difícil, y la confirmación definitiva de la enfermedad requiere confirmación molecular.
Función de los principales genes involucrados en la aparición de las ictiosis congénitas
| Gen (código MIM) | Función |
|---|---|
| Proteínas estructurales | |
| Queratinas | |
| KRT1 (139350) | Contribuye al citoesqueleto de filamentos intermedios en las células epidérmicas suprabasales. |
| KRT2 (600194) | Contribuye al citoesqueleto de filamentos intermedios en las células epidérmicas suprabasales más superficiales. |
| KRT10 (148080) | Contribuye al citoesqueleto de filamentos intermedios en las células epidérmicas suprabasales, menos importante en la piel palmoplantar. |
| Filagrina | |
| ASPRV1 (611765) | Proteasa involucrada en el procesamiento de filagrina, debido a su fenotipo, probablemente tenga más dianas. |
| CASP14 (605848) | Proteasa involucrada en el procesamiento de filagrina. |
| FLG (135940) | Proteína que agrega los filamentos intermedios de queratina a través del fomento de la formación de puentes disulfuro y la de fases separación líquido-líquido, forma parte de la envoltura cornificada y una vez proteolizada contribuye a la formación de los factores hidratantes naturales epidérmicos. |
| POMP (613386) | Chaperona molecular responsable de fomentar la formación de proteasomas y, por tanto, la maduración de proteínas críticas para la diferenciación epidérmica, como la filagrina. |
| Envoltura cornificada | |
| LORICRIN (152445) | Proteína precursora de la envoltura cornificada. |
| TGM5 (603805) | Enzima que entrecruza las proteínas precursoras de la y a la envoltura cornificada. |
| Metabolismo lipídico | |
| Ceramidas | |
| ABCA12 (607800) | Enzima involucrada en la carga de ceramidas a los cuerpos lamelares. |
| ABHD5 (604780) | Funciona como una aciltransferasa y como coactivador de la lipasa de triglicéridos en adipocitos. |
| ALDH3A2 (609523) | Enzima involucrada en la síntesis de ácidos grasos, que según algunos estudios se utilizan como precursores de ceramidas. |
| ALOX12B (603741) | Enzima involucrada en el entrecruzamiento de las ceramidas a la envoltura cornificada para formar la envoltura lipídica del corneocito. |
| ALOXE3 (607206) | Enzima involucrada en el entrecruzamiento de las ceramidas a la envoltura cornificada para formar la envoltura lipídica del corneocito. |
| CERS3 (615276) | Sintetiza ceramida a partir de ácidos grasos de cadena muy larga modificados y dihidroesfingosina. |
| CYP4F22 (611495) | ω-hidroxilación de ácidos grasos de cadena muy larga para la síntesis de acilceramida. |
| ELOVL1 (611813) | Elongasa de ácidos grasos involucrada en la síntesis de ácidos grasos de cadena muy larga, que son precursores de las ceramidas. |
| ELOVL4 (605512) | Elongasa de ácidos grasos involucrada en la síntesis de ácidos grasos de cadena muy larga, que son precursores de las ceramidas. |
| GBA1 (606463) | Cataliza la rotura de la glucosilceramida a ceramida y glucosa. |
| KDSR (136440) | Sintetiza dihidroesfingosina, precursor de las ceramidas, a partir de serina. |
| LIPN (613924) | Papel mal definido en la síntesis de ceramidas. |
| NIPAL4 (609383) | Papel mal definido en la síntesis de ceramidas. |
| PEX7 (601757) | Participa en la importación de proteínas al peroxisoma (incluyendo la fitanoil-CoA hidroxilasa). |
| PHGDH (606879) | Enzima utilizada en la síntesis de serina, que se utiliza como precursor de ceramidas. |
| PHYH (602026) | Enzima involucrada en la síntesis de ácidos grasos en el peroxisoma, que son precursores de ceramidas. |
| PNPLA1 (612121) | Cataliza la ω-O-esterificación de ácido linoleico para formar acilceramidas. |
| PSAT1 (610936) | Enzima utilizada en la síntesis de serina, que se utiliza como precursor de ceramidas. |
| PSPH (172480) | Enzima utilizada en la síntesis de serina, que se utiliza como precursor de ceramidas. |
| SDR9C7 (609769) | Enzima involucrada en el entrecruzamiento de ceramida a la envoltura cornificada para formar la envoltura lipídica del corneocito. |
| SLC27A4 (604194) | Añade coenzima A a ácidos grasos de cadena muy larga para la síntesis de ceramidas. |
| TGM1 (190195) | Enzima con funciones poco estudiadas en el entrecruzamiento de las ceramidas a la envoltura cornificada para formar la envoltura lipídica del corneocito. |
| UGCG (602874) | Glicosila acil-ceramidas. |
| Colesterol | |
| EBP (300205) | Enzima involucrada en la síntesis de colesterol. |
| MBTPS2 (300294) | Metaloproteasa de membrana involucrada en la activación de factores de transcripción relacionados con la transcripción de enzimas de la ruta biosintética del colesterol. |
| NSDHL (300275) | Enzima involucrada en la síntesis de colesterol. |
| SREBF1 (184756) | Factor de transcripción involucrado en la transcripción de enzimas de la ruta biosintética del colesterol. |
| STS (300747) | Sintetiza colesterol a partir de colesterol sulfato. |
| SULT2B1 (604125) | Responsable de la sulfonación de colesterol, que regula la diferenciación epidérmica. |
| SUMF1 (607939) | Responsable de la modificación de diferentes sulfatasas. |
| Cuerpos lamelares | |
| SNAP29 (604202) | Media eventos de fusión de cuerpos lamelares. |
| VIPAS39 (613401) | Media eventos de fusión de cuerpos lamelares. |
| VPS33B (608552) | Media eventos de fusión de cuerpos lamelares. |
| Dolicol | |
| DOLK (610746) | Fosfato dolicol. |
| MPDU1 (604041) | Añade manosa a dolicoles para la O-glicosilación y N-manosilación de proteínas. |
| PIGL (605947) | Involucrada en la síntesis de anclajes de glicosilfosfatidilinositol. |
| SRD5A3 (611715) | Involucrada en la síntesis de dolicol. |
| Uniones intercelulares | |
| Uniones ocluyentes | |
| CLDN1 (603718) | Proteína de las uniones ocluyentes, controla la permeabilidad paracelular. |
| CLDN10 (617579) | Proteína de las uniones ocluyentes, controla la permeabilidad paracelular. |
| Uniones gap | |
| GJA1 (121014) | Proteína de las uniones gap, controla la comunicación intercelular. |
| GJB2 (220290) | Proteína de las uniones gap, controla la comunicación intercelular. |
| GJB3 (603324) | Proteína de las uniones gap, controla la comunicación intercelular. |
| GJB4 (605425) | Proteína de las uniones gap, controla la comunicación intercelular. |
| GJB6 (604418) | Proteína de las uniones gap, controla la comunicación intercelular. |
| Desmosomas | |
| CDSN (602593) | Componente de los corneodesmosomas en el estrato córneo, aumenta su resistencia mecánica. |
| DSG1 (125670) | Proteína desmosómica, asegura la adhesión intercelular. |
| DSP (125647) | Proteína desmosómica, asegura la adhesión intercelular. |
| FLG2 (616284) | Asegura la adhesión intercelular en las capas superiores de la epidermis de forma dependiente de corneodesmosina. |
| Proteasas e inhibidores | |
| SPINK5 (605010) | Inhibidor de serina proteasas, impide la degradación de uniones intercelulares. |
| CAST (114090) | Inhibidor de cisteína proteasas, impide la degradación de uniones intercelulares. |
| CSTA (184600) | Inhibidor de cisteína proteasas, impide la degradación de uniones intercelulares. |
| SERPINB8 (601697) | Inhibidor de serina proteasas, impide la degradación de uniones intercelulares. |
| ST14 (606797) | Activador epitelial de membrana de otras proteasas. También tiene funciones en el procesamiento de la profilagrina y crecimiento del folículo piloso. |
| Transcripción/traducción | |
| AARS1 (601065) | Sintetasa de alanil RNA de transferencia. |
| ERCC2 (126340) | Componente del complejo TFIIH involucrado en reparación por escisión de nucleótidos y transcripción de genes tipo 2. |
| ERCC3 (133510) | Componente del complejo TFIIH involucrado en reparación por escisión de nucleótidos y transcripción de genes tipo 2. |
| GTF2E2 (189964) | Componente del complejo TFIIE involucrado en la transcripción de genes tipo 2. |
| GTF2H5 (608780) | Componente del complejo TFIIH involucrado en reparación por escisión de nucleótidos y transcripción de genes tipo 2. |
| MARS1 (156560) | Sintetasa de metionil RNA de transferencia. |
| RNF113A (300951) | Proteína de dedo RING involucrada en el procesamiento de los pre- RNA mensajero. |
| TARS1 (187790) | Sintetasa de treonil RNA de transferencia. |
| Canales de calcio | |
| TRPM4 (606936) | Canal iónico activado por calcio, asociado a regulación de la proliferación. |
| Vesículas recubiertas de clatrina | |
| AP1B1 (600157) | Parte del complejo adaptador de las vesículas recubiertas de clatrina. |
| AP1S1 (603531) | Parte del complejo adaptador de las vesículas recubiertas de clatrina. |
| Otros | |
| MPLKIP (609188) | Interacciona con quinasas dependientes de ciclina y polo quinasas, mantiene la integridad del ciclo celular. |
MIM: código del gen en la base de datos OMIM.
Según la clasificación consenso de las ictiosis congénitas publicada en 20108, las ictiosis congénitas se dividen en ictiosis no sindrómicas (cuando todos los síntomas están causados por el defecto de la barrera epidérmica y solo hay manifestaciones cutáneas) e ictiosis sindrómicas (cuando el gen causal tiene funciones extracutáneas que determinan manifestaciones en otros órganos)9. Siguiendo este mismo criterio, hemos incluido enfermedades descritas con posterioridad o que se han caracterizado mejor a lo largo de los últimos años, actualizándolas tanto clínica como molecularmente.
Ictiosis no sindrómicasSon las ictiosis cuyas alteraciones genéticas afectan solo a la diferenciación de la epidermis. Se subdividen en función de la presentación de un momento de aparición, de su patrón hereditario, su espectro fenotípico o el tipo de afectación genética (tabla 2)8.
Clasificación de las ictiosis no sindrómicas
| Ictiosis no sindrómicas |
|---|
| Ictiosis vulgar (IV) (ORPHA: −) |
| Ictiosis recesiva ligada al X (IRLX) (ORPHA: 461) |
| Ictiosis congénita autosómica recesiva (ORPHA: 281097) |
| Ictiosis laminar o lamelar (ORPHA: 313) |
| Eritrodermia ictiosiforme congénita (ORPHA: 79394) |
| Ictiosis arlequín (ORPHA: 457) |
| Bebé colodión automejorativo (ORPHA: 281122) |
| Bebé colodión automejorativo acral (ORPHA: 281127) |
| Ictiosis en traje de baño (ORPHA: 100976) |
| Ictiosis queratinopáticas (ORPHA: 281103) |
| Ictiosis epidermolítica autosómica dominante (ORPHA: 312) |
| Ictiosis epidermolítica autosómica recesiva (ORPHA: 512103) |
| Ictiosis epidermolítica anular (ORPHA: 281139) |
| Ictiosis Curth-Macklin (ORPHA: 79503) |
| Ictiosis con confeti (ORPHA: 281190) |
| Ictiosis epidermolítica superficial (ORPHA: 455) |
| Nevo epidermolítico (EN, ORPHA: 497737) |
| Síndromes de exfoliación cutánea (ORPHA: 817) |
| Síndrome de descamación cutánea generalizada (ORPHA: 263543) |
| Síndrome de descamación cutánea acral (ORPHA: 263534) |
| Síndrome PLACK (ORPHA: 44138) |
| Eritroqueratodermias (ORPHA: 79355) |
| Queratodermia por loricrina (ORPHA: 79395) |
| Síndrome KLICK (ORPHA: 281201) |
KLICK: keratosis linearis, ichthyosis congenita, and sclerosing keratoderma (queratosis linear, ictiosis congénita y queratoderma esclerosante); ORPHA: código de la enfermedad en la base de datos ORPHANET; PLACK: peeling skin, leukonychia, acral punctate keratosis, cheilitis, and knuckle pads (piel descamada, leuconiquia, keratosis puntada acral, queilitis y almohadillas en los nudillos).
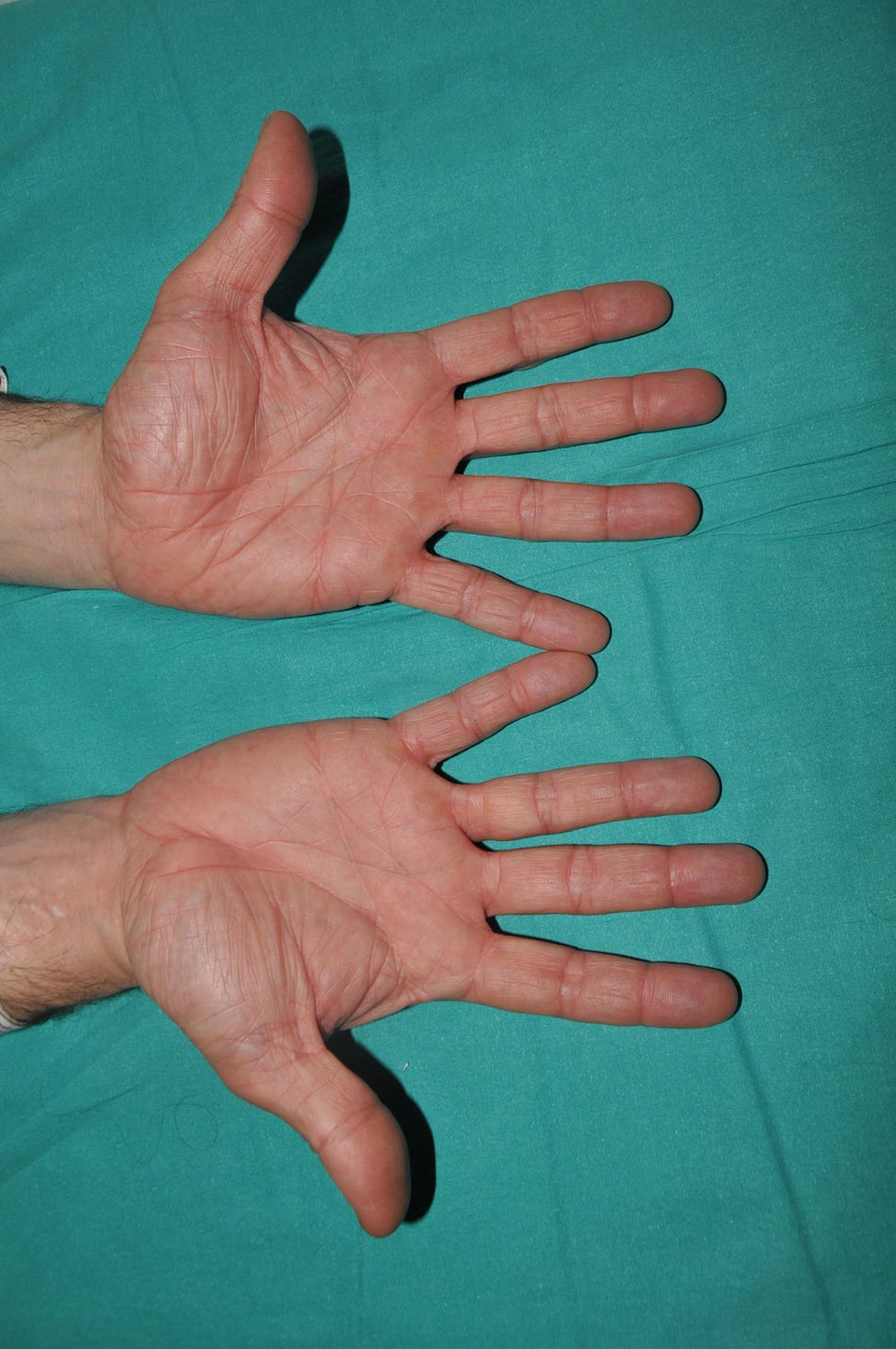
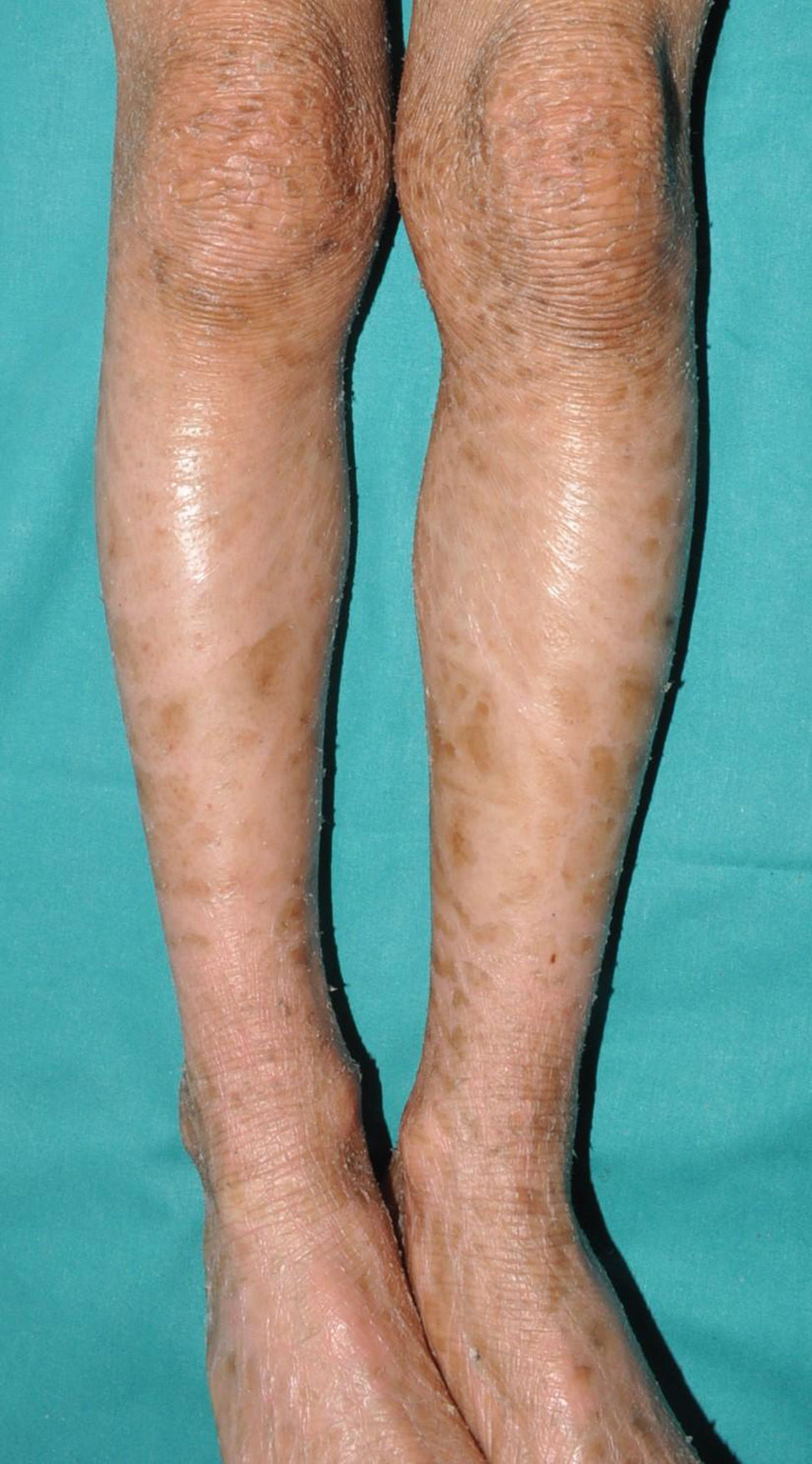
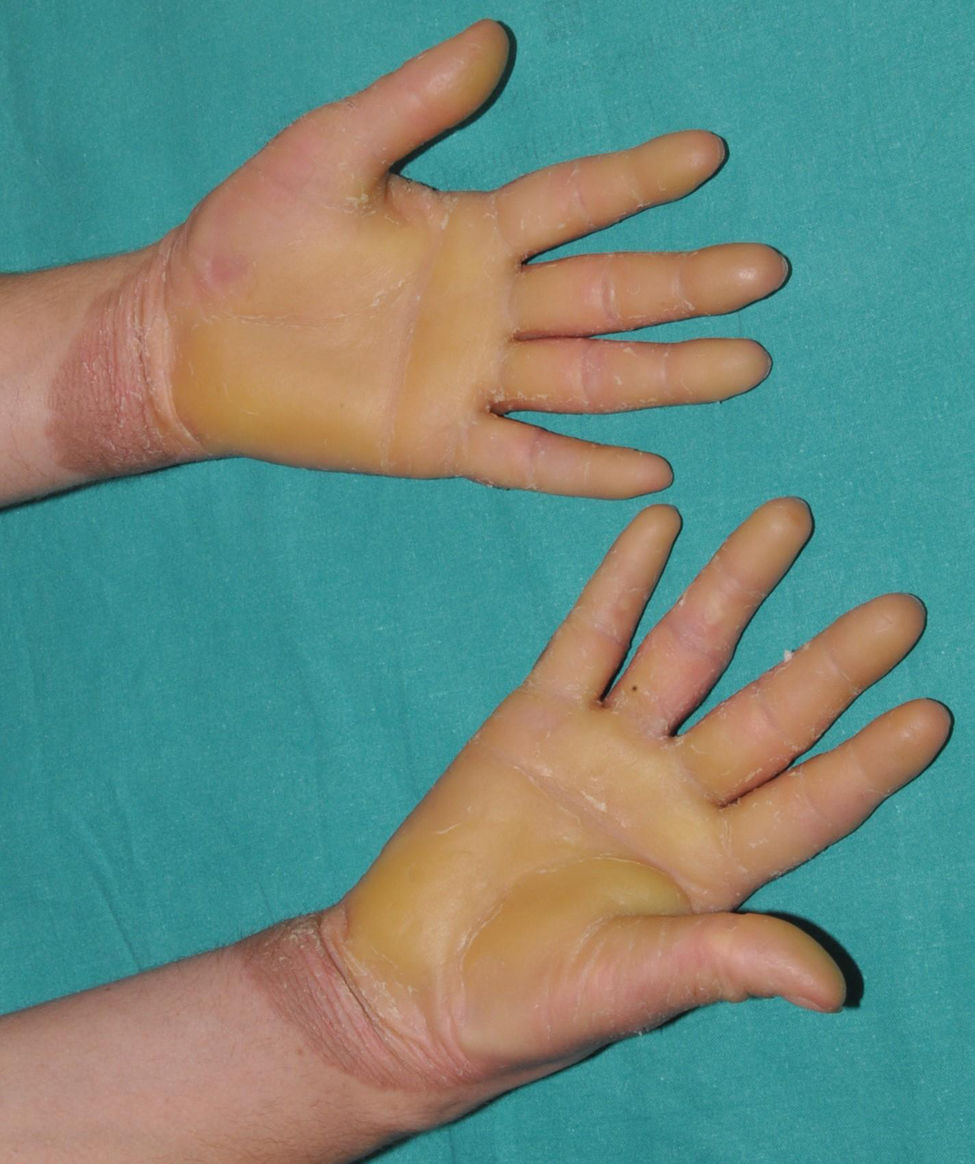
Es la forma más común de ictiosis en poblaciones norte europeas, con una prevalencia de uno en 80 en cohortes de pacientes ingleses10. La ictiosis vulgar (IV) se caracteriza, en la mayoría de los casos, por una presentación relativamente tardía, unos meses después del nacimiento. Los pacientes presentan una descamación fina y blanquecina, y aunque es más prominente en la superficie de extensión de las extremidades no es raro que afecte a toda la superficie corporal. En ocasiones, los pacientes presentan escamas de mayor tamaño y tono marrón claro en las zonas de extensión de las extremidades, sobre todo las inferiores. Otro dato característico de la IV es la hiperlinearidad palmar, hallazgo que se conoce como palma de filagrina11 (fig. 2). Con mucha menor frecuencia se observa queratodermia palmoplantar12. Además, muchos pacientes sufren picor, y el rascado determina unas lesiones lineales blanquecinas muy expresivas secundarias a un suave rascado. Aunque la IV se asocia con frecuencia a la dermatitis atópica, solo la mitad de los pacientes con esta enfermedad inflamatoria asocian variantes patogénicas en FLG13. La IV se debe a variantes patogénicas semidominantes en FLG14, gen que codifica la proteína filagrina, la proteína mayoritaria del interior de los corneocitos. Es interesante destacar que existen otras 2 formas de ictiosis mucho más raras que también afectan al metabolismo de la filagrina, originadas por variantes patogénicas recesivas en CASP1415 y dominantes en ASPRV116. Mientras que la primera presenta un fenotipo similar a la IV, la segunda, excepcional, presenta un fenotipo similar a la ictiosis laminar (ver más adelante), pero con hiperlinearidad palmar y sin membrana colodión en el nacimiento16.
Ictiosis recesiva ligada al cromosoma X en un paciente con variantes patogénicas en FLG.")
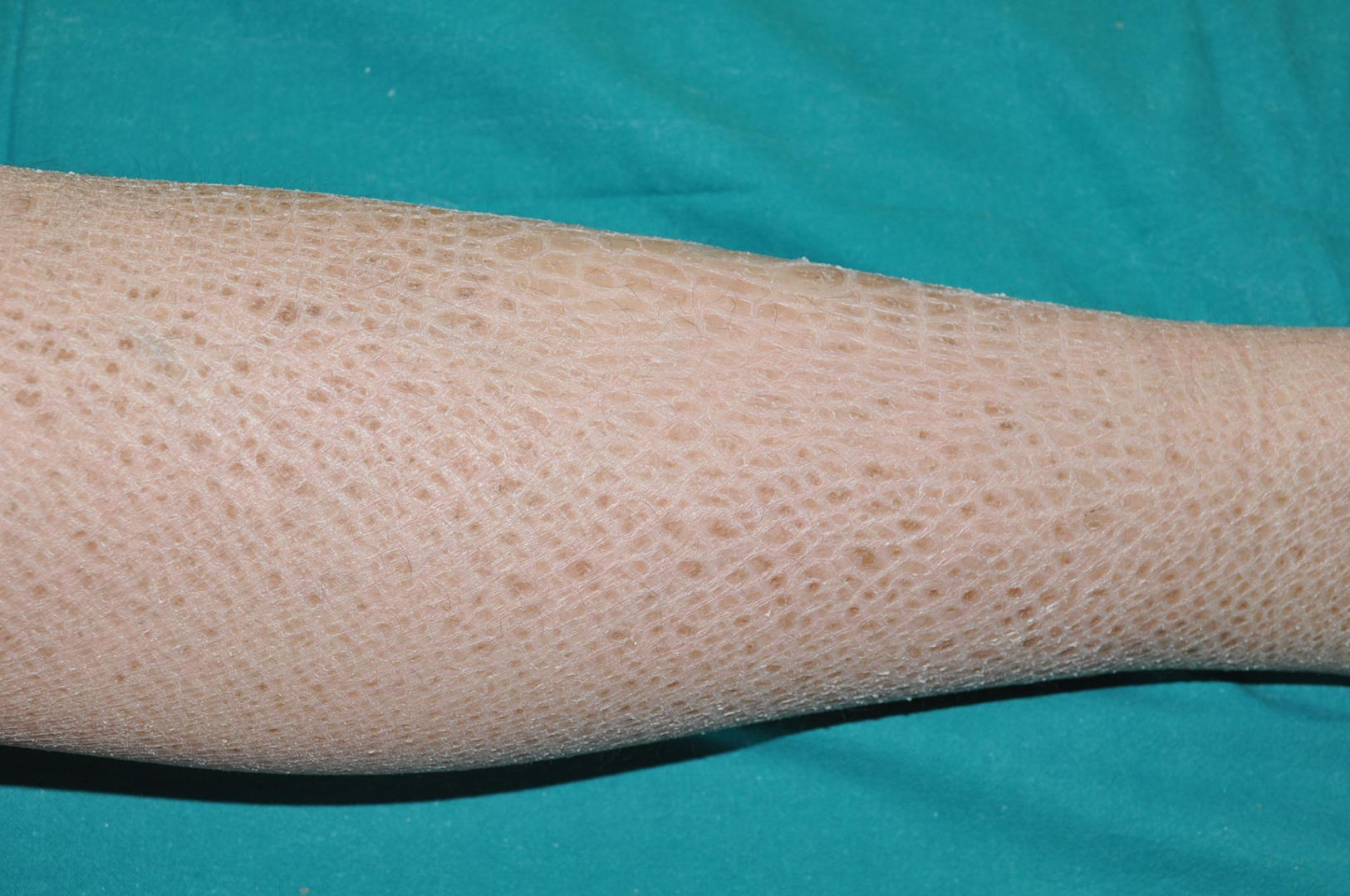
La ictiosis recesiva ligada al cromosoma (IRLX) afecta solo a los varones (con una prevalencia uno de cada 5.000)17, ya que está causada por la deleción del gen de la sulfatasa esteroidea (STS), localizado en una región distal del brazo corto del cromosoma X que no sufre inactivación18. Aunque las mujeres son portadoras de la enfermedad y la transmiten, solo la manifiestan en los casos excepcionales en los que la deleción del gen se produce en ambos cromosomas19. La IRLX se caracteriza por la presencia de escamas poligonales de tono marrón oscuro en las superficies de extensión de las extremidades (fig. 3). El tamaño de las escamas es variable, pero tienden a ser más extensas en las superficies de extensión de las extremidades inferiores. La afectación de las flexuras es variable, y probablemente depende de la severidad de la descamación, estando presente en los sujetos con mayor expresividad clínica. La descamación afecta al cuero cabelludo, el cuello, y la región retroauricular, confiriendo un falso aspecto de higiene insuficiente a algunos de estos pacientes. Las palmas y las plantas suelen estar respetadas. Como en el caso de la IV, muchos de los pacientes con IRLX nacen con la piel normal y manifiestan la enfermedad después de los primeros meses de la vida, pero algunos presentan una afectación similar (pero mucho más leve) a la membrana colodión. El déficit de función de la STS impide la desulfatación del colesterol sulfato en la epidermis, impidiendo una adecuada descamación de la capa córnea. Además, impide la desulfatación de la dehidroepiandrosterona sulfato, dificultando la maduración del cuello del útero durante el parto, provocando expulsivos prolongados en las mujeres portadoras de la enfermedad, dato clínico que puede ayudar a confirmar la sospecha clinica20. Aunque la IRLX se considera una ictiosis no sindrómica, entre el 30 y el 40% de los pacientes presentan un trastorno de déficit de atención e hiperactividad, por lo que es muy probable que algún esteroide metabolizado por el enzima STS tenga un papel como neurotransmisor21.

La IV y la IRLX pueden tener un aspecto similar en los casos de IV más sintomáticos. El despistaje de antecedentes familiares (afectación de abuelo materno o hermanos de la madre), el aspecto normal de palmas y plantas y el respeto de las flexuras en la mayoría de los pacientes con IRLX pueden ayudar a diferenciar ambas entidades. La confluencia de variantes patogénicas en FLG y STS en algunos pacientes puede aumentar de manera significativa la expresividad de la descamación en los pacientes con IRLX y facilitar que asocien hiperlinearidad palmoplantar22.
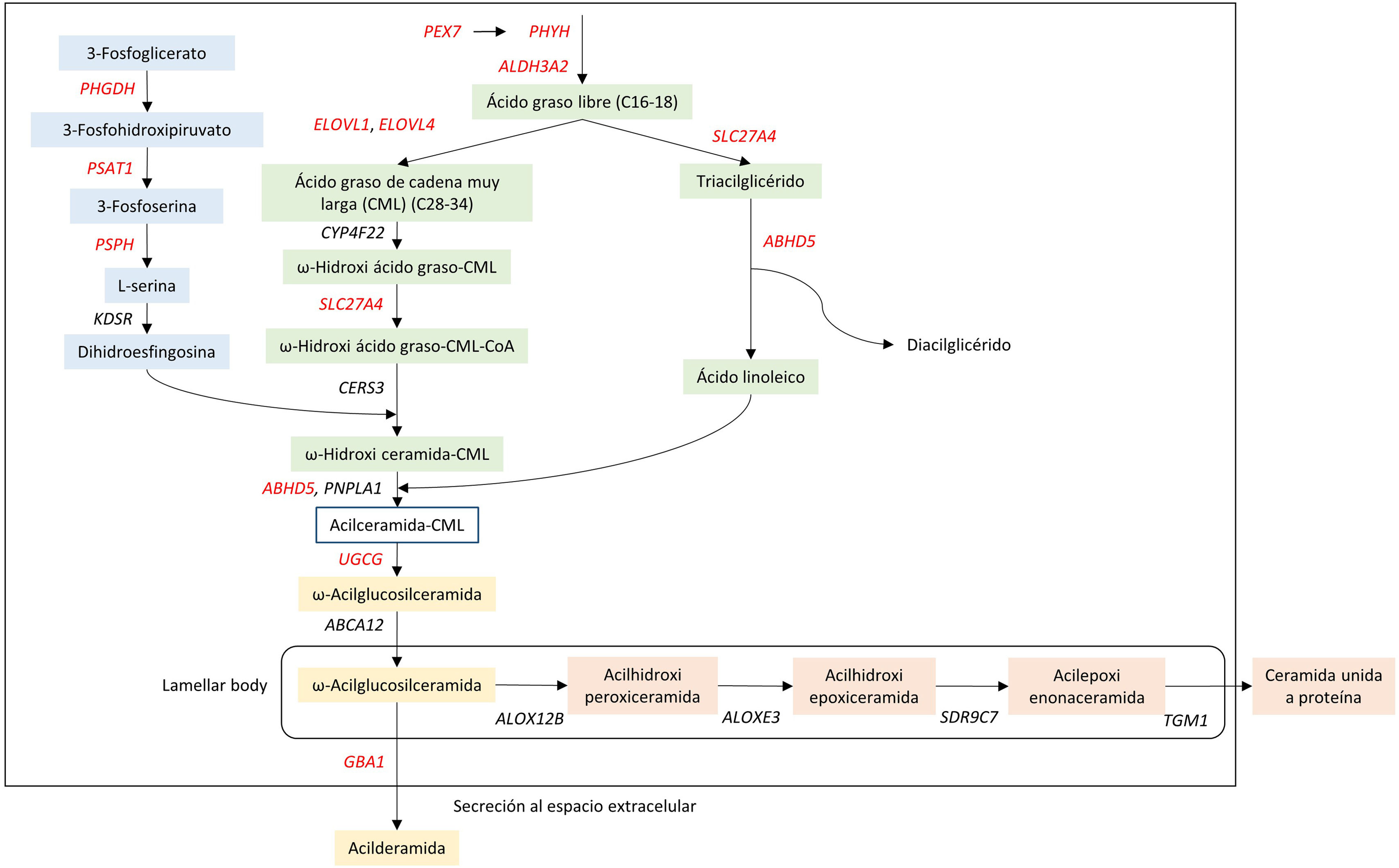
Ictiosis congénitas autosómicas recesivasLas Ictiosis congénitas autosómicas recesivas (ICAR), conocidas así por su patrón hereditario, tienen una prevalencia muy baja, estimándose entre 7,2 y 16,2 afectados por 106 habitantes tanto en España23 como en otros países del mundo24,25. A diferencia de lo que ocurre en las ictiosis del grupo anterior, las ICAR debutan en el momento del nacimiento. La forma de presentación más típica se denomina bebé colodión, que se caracteriza por una membrana brillante y tersa parecida al pegamento transparente que recubre todo el cuerpo y que, en los casos más severos, ocasiona ectropión (eversión de los párpados) y eclabión (eversión de los labios). La membrana colodión se desprende en las primeras semanas de vida, evolucionando a una presentación fenotípica de gravedad variable, con distintos grados de eritema y diferente morfología de la descamación. En función de ello, se distinguen 5 formas clínicas distintas que se detallan más abajo. En los casos en los que no se aprecia membrana colodión, los neonatos presentan eritrodermia y distintos grados de hiperqueratosis y descamación. En la mayoría de los casos, los genes afectados están involucrados en el metabolismo de ceramidas8, un componente lipídico de la matriz intercelular que evita la evaporación transepidérmica de agua (fig. 4). (tabla 3). Aunque no existe una correlación genofenotípica exacta, los pacientes con variantes patogénicas en TGM1 asocian alopecia, ectropión y presentación neonatal como bebé colodión con una frecuencia significativamente mayor que el resto de los genes8.
 y ácido graso (verde) para formar acil-ceramida que puede liberarse como lípido libre (amarillo) o unido a proteínas (naranja). Mutaciones en los genes representados causan ictiosis no sindrómicas (negro) o sindrómicas (rojo). Fuente: modificado de «Ichthyosis», C. Gutiérrez-Cerrajero et al.9.")
Ruta de síntesis de ceramidas en la epidermis. Estas reacciones constan de la unión de una base esfingoide (azul) y ácido graso (verde) para formar acil-ceramida que puede liberarse como lípido libre (amarillo) o unido a proteínas (naranja). Mutaciones en los genes representados causan ictiosis no sindrómicas (negro) o sindrómicas (rojo). Fuente: modificado de «Ichthyosis», C. Gutiérrez-Cerrajero et al.9.
Formas infrecuentes de las ictiosis no sindrómicas
| Grupo | Genes causales | Características principales |
|---|---|---|
| Síndromes de descamación cutánea (PSS, ORPHA: 817) | Caracterizados por descamación en la capa superior de la epidermis1. Se subdividen dependiendo de las áreas afectadas | |
| Síndrome de descamación cutánea generalizada (PSS generalizado, ORPHA: 263543) | CDSN2 (AR, MIM: 270300), FLG21 (AR, MIM: 618084) | La descamación afecta a la superficie cutánea entera1 e incluye 2 subtipos:El subtipo A (no inflamatorio, ORPHA: 263548) causado por FLG2.El subtipo B (inflamatorio, ORPHA: 263553) causado por CDSN. |
| Síndrome de descamación cutánea acral (PSS acral, ORPHA: 263534) | CSTA3 (AR, MIM: 607936), TGM54 (AR, MIM: 609796) | La descamación afecta primariamente a las superficies plantares y dorsales de manos y pies1. |
| Ictiosis exfoliativa (ORPHA: 289586) | CSTA5 (AR, MIM: 607936), SERPINB86 (AR, MIM: 617115) | Caracterizada por descamación de la piel y piel seca y escamosa generalizada5. No se clasifica típicamente como PSS, pero la ictiosis exfoliativa comparte síntomas y alteraciones moleculares subyacentes con PSS acral5. |
| Síndrome de descamación cutánea - leuconiquia - queratosis punctata acral - queilitis - almohadillas de nudillo (PLACK, ORPHA: 444138) | CAST7 (AR, MIM: 616295) | Caracterizado por descamación cutánea generalizada con leuconiquia (descoloración blanca de las uñas), queratosis punctata acral (parches queratósicos en las extremidades), queilitis y almohadillas en los nudillos. |
| Otros | ||
| Queratodermia loricrina (LK, ORPHA: 79395) | LORICRIN8 (AD, MIM: 604117) | También conocida como queratodermia hereditaria mutilante con ictiosis, enfermedad en camisa o síndrome de Vohwinkel con ictiosis. Se caracteriza por ictiosis generalizada, con hiperqueratosis palmoplantar en pana y a menudo bandas constrictoras alrededor de los meñiques8. |
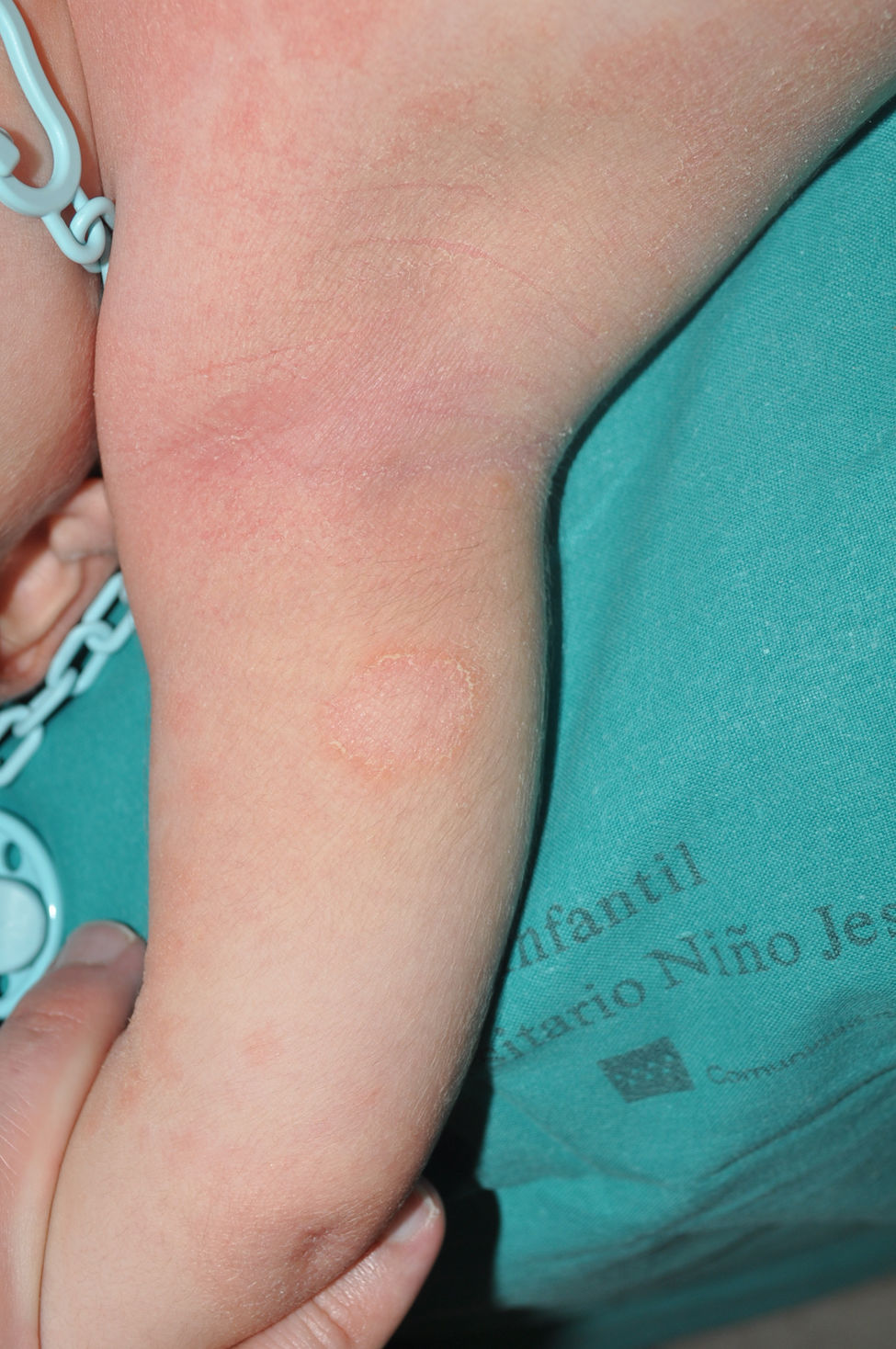
| Eritroqueratodermia variable progresiva (EKVP, ORPHA: 308166) | GJA19 (AD, MIM: 617525), GJB310 (AD o AR, MIM: 133200), GJB411 (AD, MIM: 617524), KDSR12 (AR, MIM: 617526), PERP13 (AR, MIM: 619209), TRPM414 (AD, MIM: 618531) | Un término que engloba a pacientes con hallazgos clínicos similares: eritema migratorio y lesiones hiperqueratósicas, que cambian de tamaño con el tiempo15 (a veces llamado eritroqueratodermia variable [EKV]) y/o placas fijas marrones a rojas hiperqueratóticas11 (a veces llamada eritroqueratodermia simétrica progresiva [PSEK]). Los individuos y familias pueden mostrar tanto placas fijas como migratorias causadas por mutaciones en diferentes genes, algunos de los cuales codifican proteínas sin aparente relación funcional. Las características de EKVP a veces se han descrito en algunos pacientes con mutaciones en NIPAL416 o ABCA1217. |
| Síndrome de queratosis linear - ictiosis congénita - queratodermia esclerosante (KLICK, ORPHA: 281201) | POMP18 (AR, MIM: 601952) | Caracterizado por ictiosis congénita, pápulas discretas en los aspectos flexurales de las articulaciones, queratodermia palmoplantar, bandas constrictoras alrededor de los dedos y deformidades flexurales18. |
AD: autosómico dominante; AR: autosómico recesivo.
1. Alfares A, Al-Khenaizan S, Al Mutairi F, Fuad C, Mutairi A, Abdullah K. Peeling skin syndrome associated with novel variant in FLG2 gene. Am J Med Genet A. 2017;173;3201-3204. https://doi.org/10.1002/AJMG.A.38468.
22. Oji V, Eckl KM, Aufenvenne K, Nätebus M, Tarinski T, Ackermann K, et al. Loss of Corneodesmosin Leads to Severe Skin Barrier Defect, Pruritus, and Atopy: Unraveling the Peeling Skin Disease. Am J Hum Genet. 2010;87;274-281. https://doi.org/10.1016/J.AJHG.2010.07.005.
3. Muttardi K, Nitoiu D, Kelsell DP, O’Toole EA, Batta K. Acral peeling skin syndrome associated with a novel CSTA gene mutation. Clin Exp Dermatol. 2016;41;394–398. https://doi.org/10.1111/CED.12777.
4. Cassidy AJ, van Steensel MAM, Steijlen PM, van Geel M, van der Velden J, Morley SM, et al. A homozygous missense mutation in TGM5 abolishes epidermal transglutaminase 5 activity and causes acral peeling skin syndrome. Am J Hum Genet. 2005;77;909-917. https://doi.org/10.1086/497707.
5. Blaydon DC, Nitoiu D, Eckl KM, Cabral RM, Bland P, Hausser I, et al. Mutations in CSTA, encoding Cystatin A, underlie exfoliative ichthyosis and reveal a role for this protease inhibitor in cell-cell adhesion. Am J Hum Genet. 2011;89;564-571. https://doi.org/10.1016/J.AJHG.2011.09.001.
6. Pigors M, Sarig O, Heinz L, Plagnol V, Fischer J, Mohamad J, et al. Loss-of-Function Mutations in SERPINB8 Linked to Exfoliative Ichthyosis with Impaired Mechanical Stability of Intercellular Adhesions. Am J Hum Genet. 2016;99;430-436. https://doi.org/10.1016/J.AJHG.2016.06.004.
7. Lin Z, Zhao J, Nitoiu D, Scott CA, Plagnol V, Smith FJD, et al. Loss-of-function mutations in CAST cause peeling skin, leukonychia, acral punctate keratoses, cheilitis, and knuckle pads. Am J Hum Genet. 2015;96;440-447. https://doi.org/10.1016/J.AJHG.2014.12.026.
8. Maestrini E, Monaco AP, McGrath JA, Ishida-Yamamoto A, Camisa C, Hovnanian A, et al. A molecular defect in loricrin, the major component of the cornified cell envelope, underlies Vohwinkel's syndrome. Nat Genet. 1996;13;70-77. https://doi.org/10.1038/ng0596-70.
9. Boyden LM, Craiglow BG, Zhou J, Hu R, Loring EC, Morel KD, et al. Dominant De Novo Mutations in GJA1 Cause Erythrokeratodermia Variabilis et Progressiva, without Features of Oculodentodigital Dysplasia. J Invest Dermatol. 2015;135;1540-1547. https://doi.org/10.1038/JID.2014.485.
10. Richard G, Smith LE, Bailey RA, Itin P, Hohl D, Epstein EH, et al. Mutations in the human connexin gene GJB3 cause erythrokeratodermia variabilis. Nat Genet. 1998;20;366-369. https://doi.org/10.1038/3840.
11. Macari F, Landau M, Cousin P, Mevorah B, Brenner S, Panizzon R, et al. Mutation in the Gene for Connexin 30.3 in a Family with Erythrokeratodermia Variabilis. Am J Hum Genet. 2000;67;1296-1301. https://doi.org/10.1016/S0002-9297(07)62957-7.
12. Boyden LM, Vincent NG, Zhou J, Hu R, Craiglow BG, Bayliss SJ, et al. Mutations in KDSR Cause Recessive Progressive Symmetric Erythrokeratoderma. Am J Hum Genet. 2017;100;978-984. https://doi.org/10.1016/J.AJHG.2017.05.003.
13. Duchatelet S, Boyden LM, Ishida-Yamamoto A, Zhou J, Guibbal L, Hu R, et al. Mutations in PERP Cause Dominant and Recessive Keratoderma. J Invest Dermatol. 2019;139;380-390. https://doi.org/10.1016/J.JID.2018.08.026.
14. Wang H, Xu Z, Lee BH, Vu S, Hu L, Lee M, et al. Gain-of-Function Mutations in TRPM4 Activation Gate Cause Progressive Symmetric Erythrokeratodermia. J Invest Dermatol. 2019;139;1089-1097. https://doi.org/10.1016/J.JID.2018.10.044/ATTACHMENT/4A5430A4-C020-471F-B7C0-96992C1474D1/MMC1.PDF.
15. Van Steensel MAM, Oranje AP, van der Schroeff JG, Wagner A, van Geel M. The missense mutation G12D in connexin30.3 can cause both erythrokeratodermia variabilis of Mendes da Costa and progressive symmetric erythrokeratodermia of Gottron. Am J Med Genet A. 2009;149A;657-661. https://doi.org/10.1002/AJMG.A.32744.
16. Boyden LM, Craiglow BG, Hu RH, Zhou J, Browning J, Eichenfield L, et al. Phenotypic spectrum of autosomal recessive congenital ichthyosis due to PNPLA1 mutation. Br J Dermatol. 2017;177;319-322. https://doi.org/10.1111/BJD.15570.
17. Sun Q, Burgren NM, Cheraghlou S, Paller AS, Larralde M, Bercovitch L, et al. The Genomic and Phenotypic Landscape of Ichthyosis: An Analysis of 1000 Kindreds. JAMA Dermatol. 2022;158;16-25. https://doi.org/10.1001/JAMADERMATOL.2021.4242.
18. Dahlqvist J, Klar J, Tiwari N, Schuster J, Törmä H, Badhai J, et al. A single-nucleotide deletion in the POMP 5’ UTR causes a transcriptional switch and altered epidermal proteasome distribution in KLICK genodermatosis. Am J Hum Genet. 2010;86;596-603. https://doi.org/10.1016/J.AJHG.2010.02.018.
Según la clasificación vigente de las ictiosis8, existen 5 formas clínicas de ICAR, que incluyen:
- –
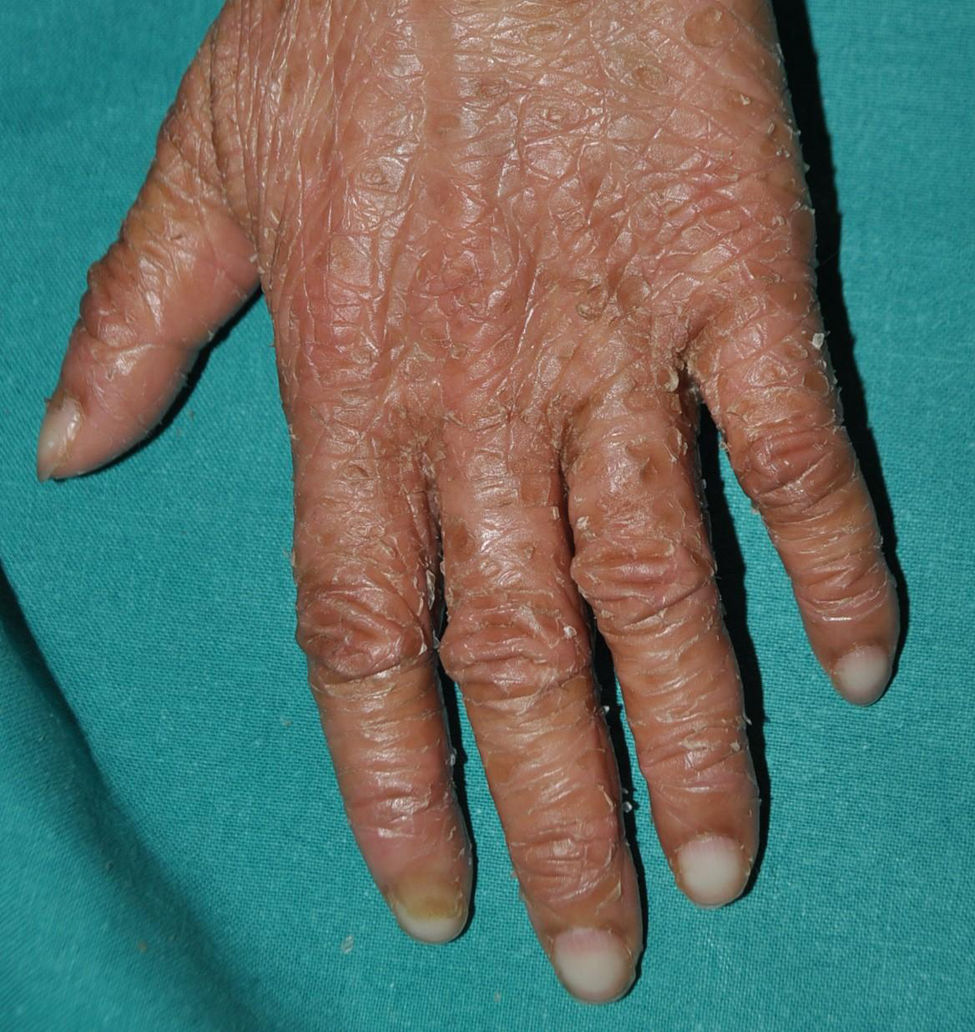
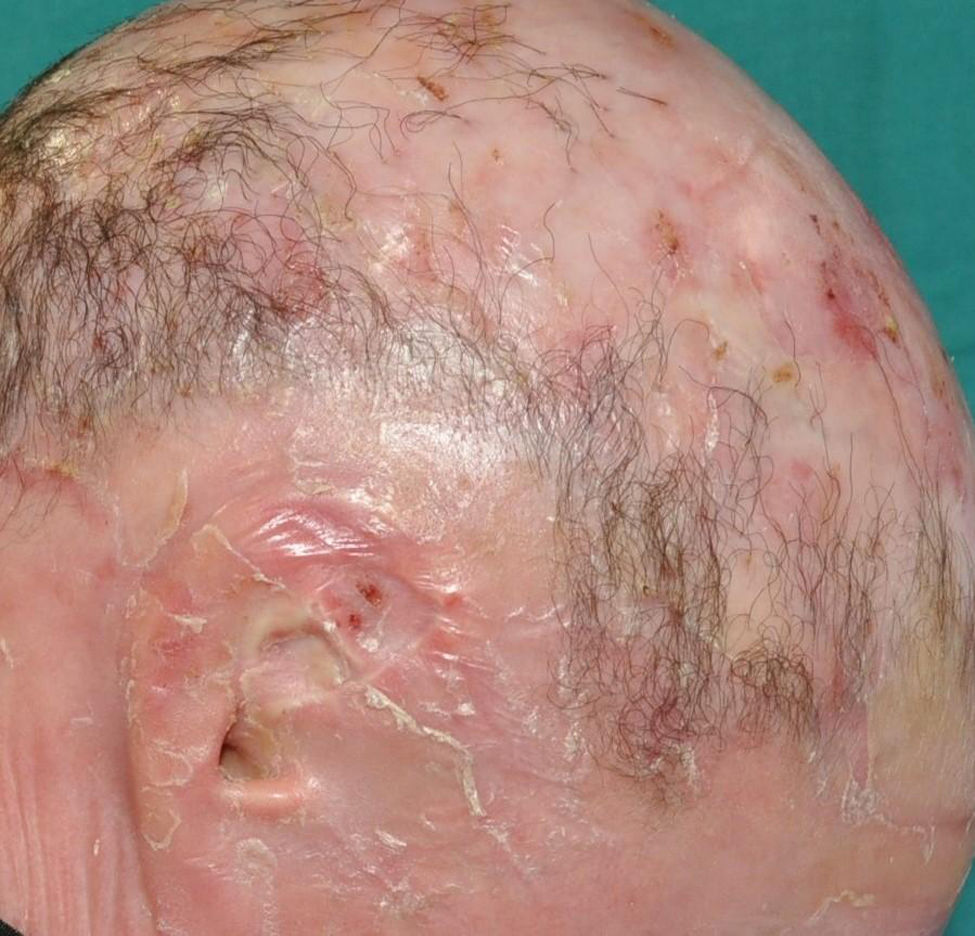
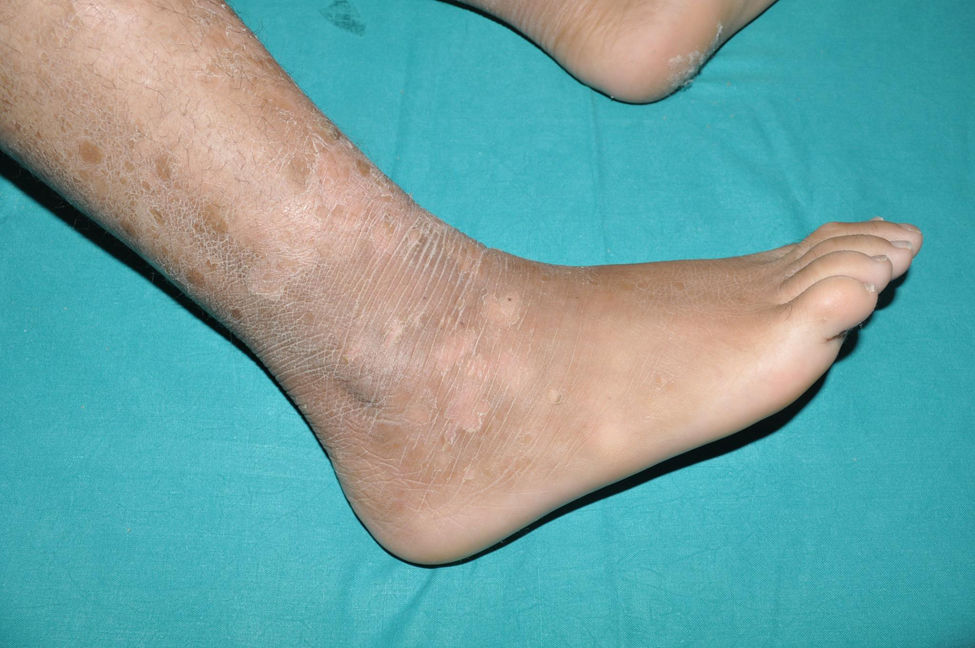
Ictiosis laminar o lamelar: se caracteriza por un grado moderado a intenso de eritema y descamación en grandes láminas de tono algo más oscuro que el de la piel del paciente, es decir, marrón claro en fototipos claros y marrón más oscuro es fototipos más oscuros. Esta descamación grande a modo de láminas afecta a todo el cuerpo, pero es más evidente en las extremidades inferiores y la región frontal de la cara, siendo notablemente más oscura en los fototipos altos (fig. 5). Muchos pacientes presentan, además, ectropión y una descamación gruesa adherida en el cuero cabelludo que puede anteceder a una alopecia cicatricial prácticamente total26. La afectación palmoplantar también presenta un aspecto laminar, con áreas de engrosamiento difuso de superficie lisa (fig. 6). No es infrecuente la existencia de alteraciones de la morfología ungueal, como la incurvación de la lámina ungueal, áreas lineales de leuconiquia y aumento de la extensión de la lúnula.
- –
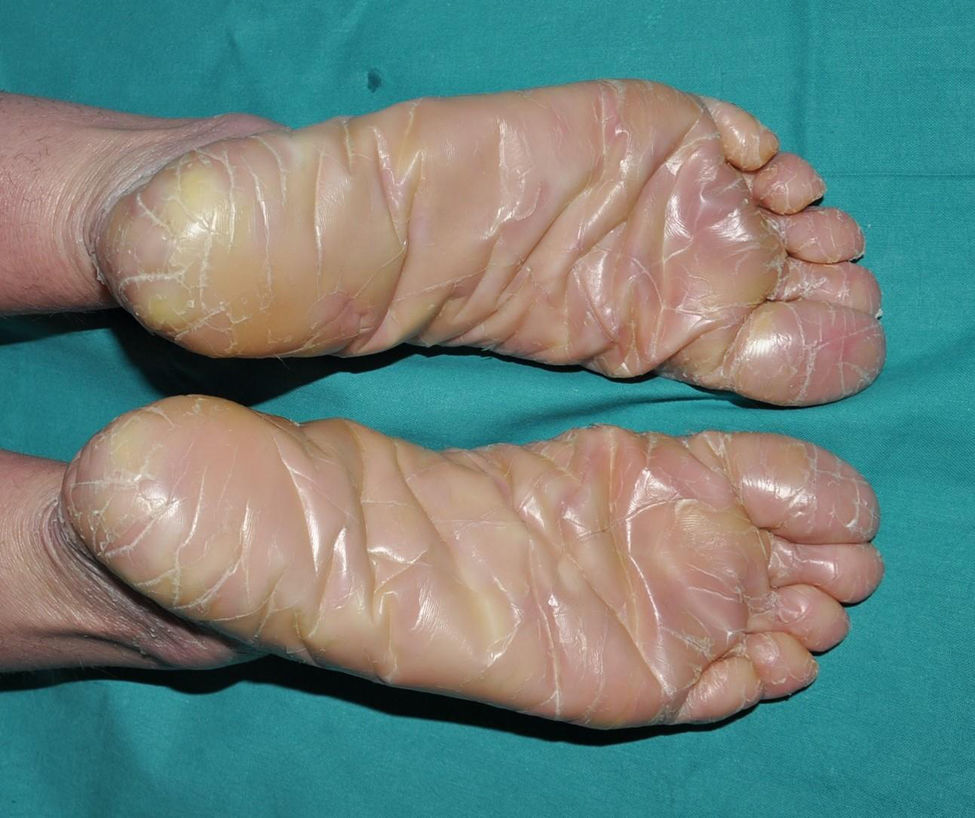
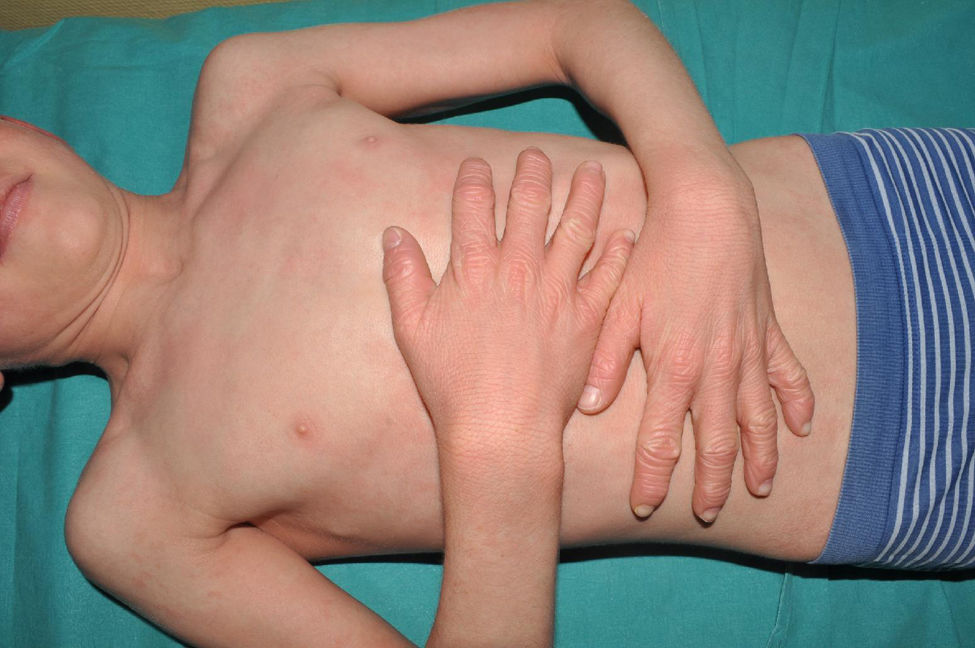
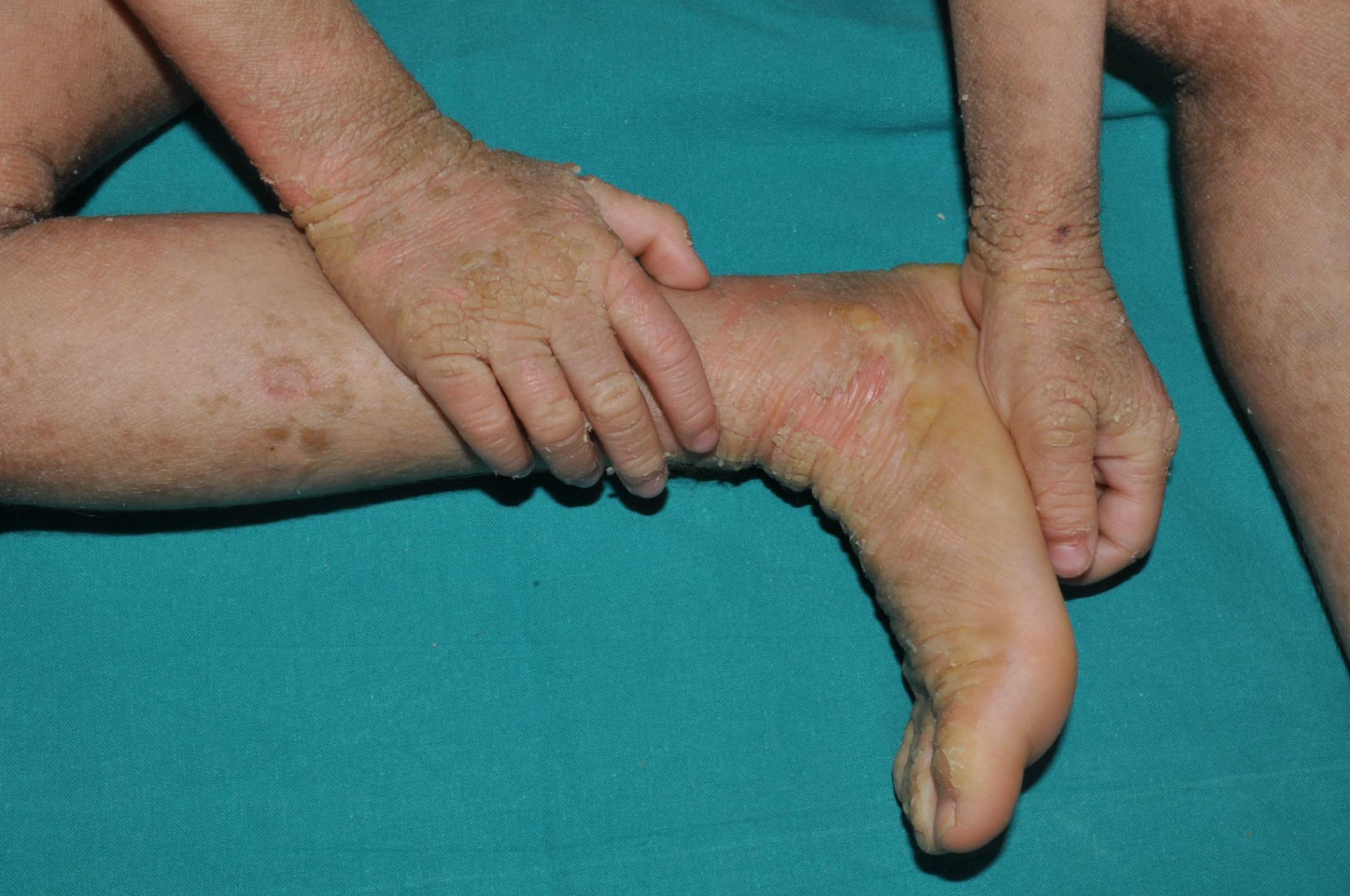
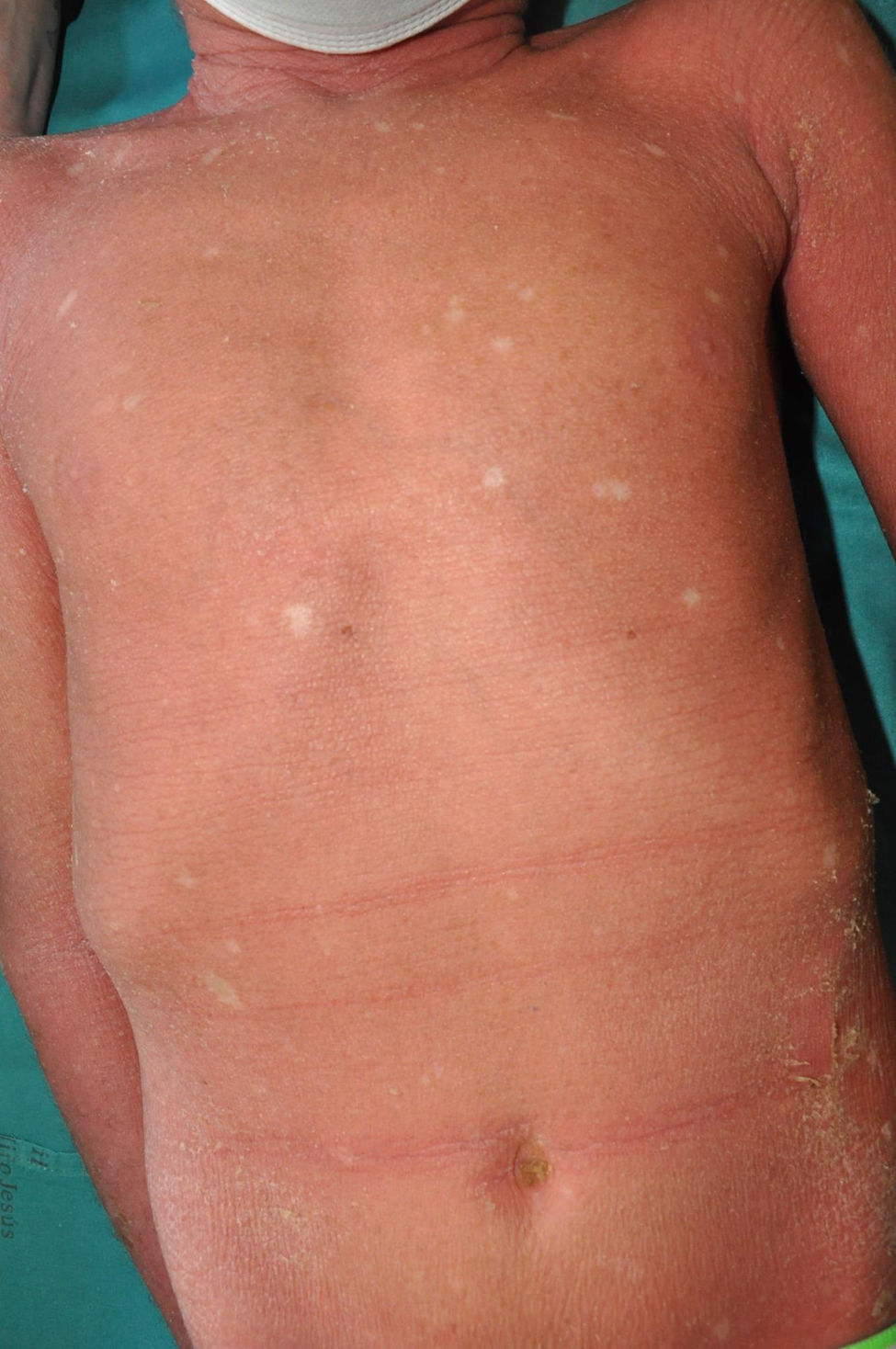
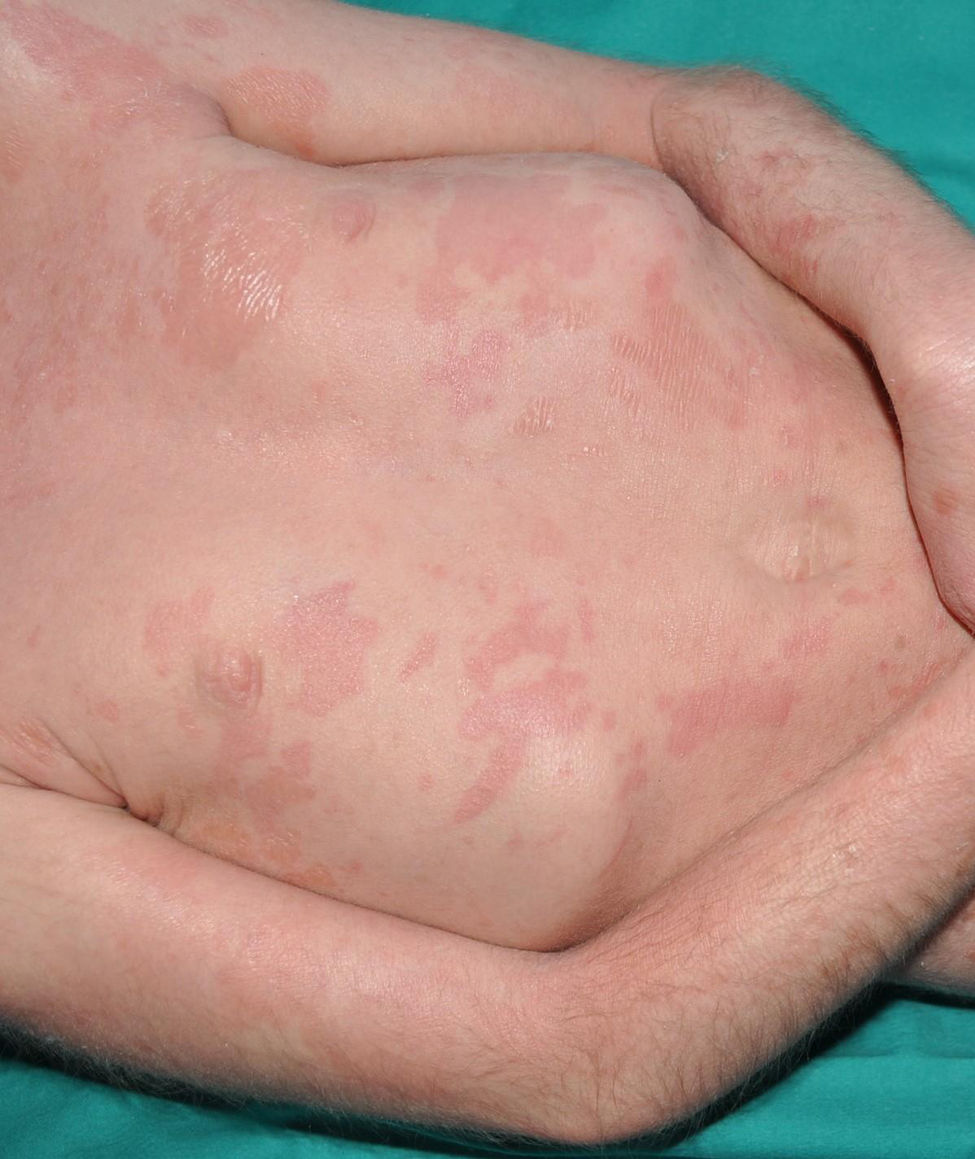
Eritrodermia ictiosiforme congénita: es, junto con la ictiosis laminar, un fenotipo clásico de las ICAR. Los pacientes presentan una eritrodermia de variable intensidad y descamación generalizada con escamas más pequeñas que las de las formas laminares (fig. 7). El cuero cabelludo presenta una descamación habitualmente menos gruesa y adherida que en las formas laminares. Es característico que los pacientes presenten engrosamiento cutáneo del dorso de las manos y los pies y una relativa pérdida de elasticidad de la piel de los dedos que determina una cierta retracción de los mismos y un fenómeno de seudoleuconiquia al estirarlos, ya que el lecho cutáneo queda exangüe (fig. 8). Además, muchos pacientes presentan también una leuconiquia verdadera de probable origen postinflamatorio. La afectación palmoplantar determina un engrosamiento de las palmas y las plantas sin verdadera hiperqueratosis de aspecto similar al cuero (fig. 9). Muchos pacientes presentan, además, un llamativo enrojecimiento facial en las mejillas independiente de la gravedad de afectación del resto de la superficie corporal.
- –
Ictiosis arlequín: es la forma grave de las ICAR. Recibe es nombre porque los bebés nacen con una piel rígida y engrosada con fisuras lineales extensas y profundas que recuerdan al traje de los arlequines. La rigidez del tegumento cutáneo es tal que restringe los movimientos respiratorios y la capacidad de succión, poniendo en peligro la vida del niño. Así mismo, los pacientes presentan ectropión severo, eclabium, y una limitación grave de la movilidad de los dedos de las manos, que quedan englobados por la coraza córnea. Este fenotipo evoluciona semanas más tarde a una forma grave de eritrodermia ictiosiforme congénita en la que se asocian, entre otros síntomas, alopecia de cuero cabelludo, cejas y pestañas, ectropión, anomalías de los pabellones auriculares, deformidades articulares, y limitaciones permanentes de movilidad27 (fig. 10). Está causada por variantes patogénicas recesivas que causan pérdida de función completa de ABCA1227, con fenotipos ligeramente menos graves en pacientes con 2 variantes patogénicas heterocigotas diferentes28.
- –
Bebé colodión automejorativo: es una variante clínica en la que la membrana colodión evoluciona, independientemente de su gravedad, a un fenotipo leve de ICAR. Aunque la expresividad de la ictiosis es muy tenue en algunas zonas como el tronco y la parte proximal de las extremidades, los pacientes suelen presentar eritema facial, el engrosamiento cutáneo en codos, rodillas, y dorso de manos y pies (fig. 11), así como engrosamiento palmoplantar de aspecto similar al descrito en la eritrodermia ictiosiforme congénita. La evolución fenotípica hacia las formas automejorativas no es predecible, lo cual dificulta la información pronóstica de los bebés colodión en los primeros días de la vida. De hecho, está causado por variantes patogénicas recesivas en ALOX12B29, ALOXE330, CYP4F2231 o TGM132, genes que también pueden determinar una afectación clínica mucho más importante y, por tanto, ni siquiera un diagnóstico molecular inmediato puede predecir la evolución.
- –
Bebé colodión automejorativo acral es una forma clínica excepcional en la que la membrana colodión solo afecta a la parte distal de las extremidades. Está causado por variantes patogénicas recesivas en TGM133.
- –
Ictiosis en traje de baño es la una variante de las ICAR en la que los pacientes nacen con membrana colodión y evolucionan a un fenotipo de ictiosis laminar de variable expresividad que solo afecta al tronco y el cuero cabelludo. Es una forma excepcional inicialmente descrita en población norteafricana34, y está causada por variantes patogénicas recesivas en TGM135 que generan una proteína termosensible que pierde actividad en las zonas más calientes del cuerpo.







Las ictiosis queratinopáticas son denominadas así porque están causadas por variantes patogénicas en genes que codifican queratinas. Tienen una prevalencia muy baja, estimada en 1,1 por millón24. En función del patrón hereditario y los rasgos fenotípicos se distinguen distintas formas clínicas:
- –
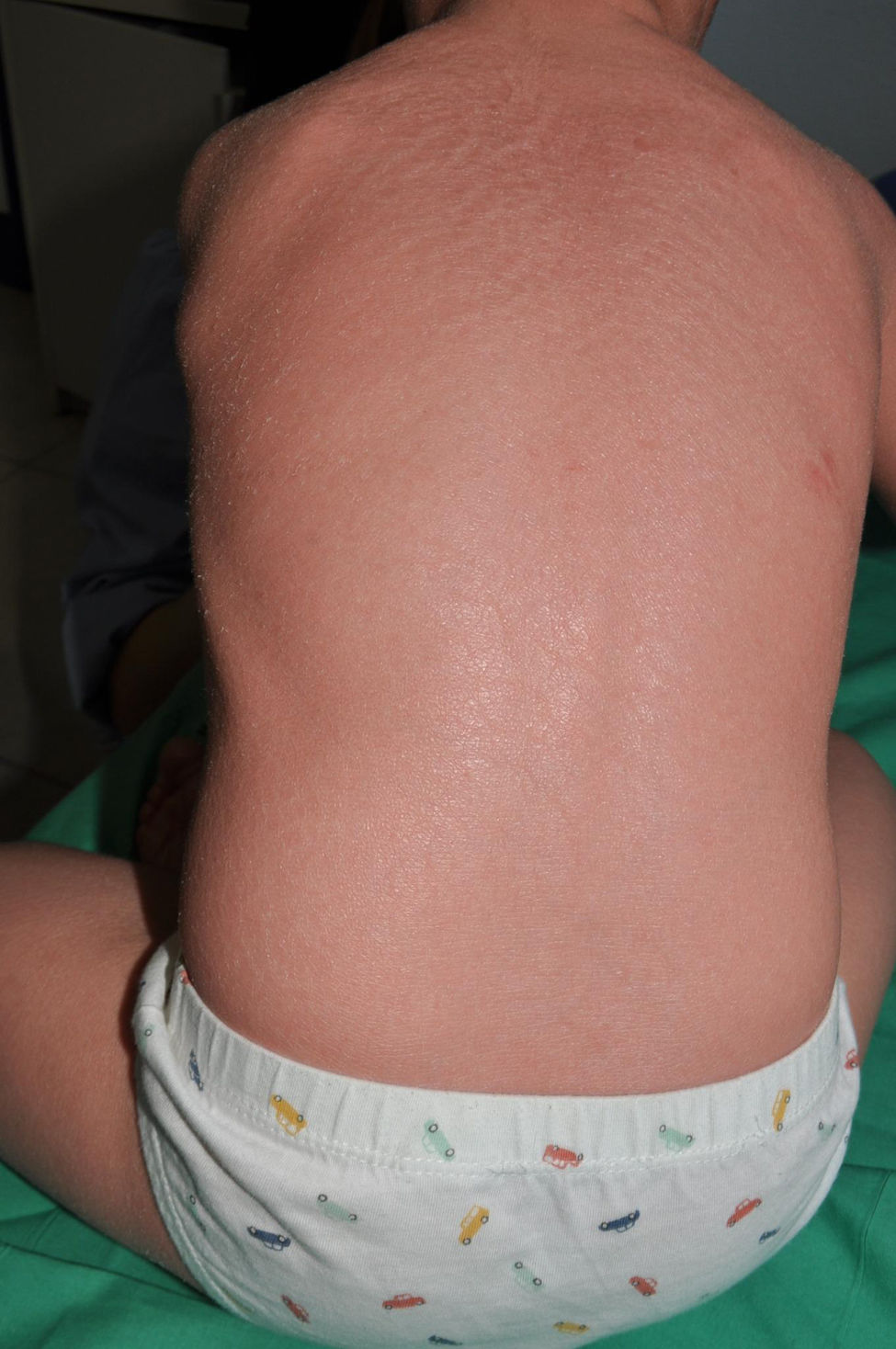
Ictiosis queratinopática autosómica dominante: es la forma más común de las ictiosis queratinopáticas, y su característica fundamental es la separación intercelular, por lo que se las denomina también ictiosis epidermolítica. Los pacientes nacen con eritrodermia, ampollas y erosiones tan extensas que pueden dificultar el diagnóstico diferencial con las epidermólisis ampollosas. Con el paso de las semanas, los pacientes sufren menos erosiones y se desarrolla una hiperqueratosis difusa que se irá acentuando progresivamente en las superficies flexoras de los grandes pliegues y, posteriormente, en codos y rodillas. Los pacientes presentan a lo largo de la vida un grado variable de eritema y de fragilidad cutánea que les hace susceptibles a las erosiones ante mínimos traumatismos (fig. 12). Este grupo de ictiosis está causado por variantes dominantes en KRT136 o KRT1036. Las causadas por variantes en KRT1 suelen presentar queratodermia palmoplantar (fig. 13), habitualmente ausente en las asociadas a variantes patogénicas en KRT10, dado que en piel palmoplantar la KRT9 puede rescatar parcialmente el fenotipo de una pérdida de KRT1036.
 Figura 12.
Figura 12.Ictiosis queratinopática epidermolítica por variante patogénica en gen KRT10. Se observa una afectación generalizada con áreas hiperqueratósicas en extremidades superiores e inferiores y áreas de fragilidad cutánea en región pretibial y lateral de los pies. La afectación se detiene en el borde palmoplantar y respeta palmas y plantas.
(0.28MB). - –
Ictiosis queratinopática autosómica recesiva: clínicamente muy similar a la anterior, está causada por variantes patogénicas recesivas en KRT1037. Su prevalencia es mucho más baja que la de las formas dominantes.
- –
Ictiosis queratinopática anular: es un subtipo especial en la que los pacientes nacen con aspecto similar al de las otras ictiosis queratinopáticas, pero posteriormente evolucionan a una forma clínica menos grave en la que, además de una hiperqueratosis difusa a nivel de los grandes pliegues, se observan lesiones anulares transitorias con descamación superficial38 (fig. 14). Está causada por variantes patogénicas dominantes en KRT139 o KRT1040.
- –
Ictiosis queratinopática superficial muestra un fenotipo más leve que las 2 anteriores y no presenta eritrodermia al nacer. Se caracteriza por una hiperqueratosis difusa leve o moderada que muestra predilección por las superficies articulares. Puede haber desprendimiento focal de la hiperqueratosis, fenómeno denominado «mauserung» o «molting» en la literatura internacional y que pudiera denominarse «desconchado» en castellano (fig. 15). Está causada por variantes patogénicas dominantes en KRT241. Esta queratina se expresa en capas superficiales del estrato córneo y probablemente por ello la manifestación clínica y los fenómenos de epidermólisis y desprendimiento focal de la hiperqueratosis son menos expresivos que en los cuadros relacionados con variantes patogénicas de KRT1 y KRT10.
- –
Ictiosis queratinopáticas mosaicas se caracterizan por lesiones hiperqueratósicas de distribución Blaschkoide cuya extensión es variable, oscilando entre unos pocos centímetros (nevo epidermolítico) y la afectación hemicorporal, uni o bilateral. Están causada por variantes patogénicas poscigóticas en KRT142, KRT1043 o KRT244, en los cuales la mutación causante de la enfermedad afecta solo a algunos progenitores epidérmicos. El área de piel afectada varía en función del momento de la mutación, siendo más extensa cuanto más precoz. En los casos en los que el mosaico afecta a las gónadas, la enfermedad se puede transmitir a los hijos del paciente como ictiosis queratinopática generalizada.
- –
Ictiosis queratinopática con parches de reversión (en confetti): es un tipo particular de ictiosis queratinopática no epidermolítica en la que no existe epidermólisis y, por tanto, no se observan los hallazgos histológicos típicos de separación intercelular. Clínicamente los pacientes presentan eritrodermia ictiosiforme severa y una afectación palmoplantar variable. En la edad escolar empiezan a observarse pequeñas zonas lenticulares de piel sana en el tronco, las cuales aumentan en número y tamaño con la edad (fig. 16). Los pacientes pueden, además, presentar ectropión, alopecia de cuero cabelludo, alteraciones ungueales, hipertricosis y marcha anormal, entre otros síntomas45. Está causada por variantes dominantes en KRT1046 o, con mucha menos frecuencia, en KRT1, cuya clínica de reversión aparece en la edad adulta47. Los parches de piel sana están causados por pérdida de heterocigosidad del alelo mutado debido a eventos de recombinación mitótica cuyo factor desencadenante es desconocido, pero se cree que pueden tener relación con la importación aberrante de las queratinas mutadas al núcleo45.
- –
Otras ictiosis no sindrómicas se detallan en la tabla 3. Son muy poco frecuentes, pero algunas de ellas tienen datos clínicos prominentes que pueden facilitar el diagnóstico, como la descamación superficial y el eritema subyacente en los síndromes de exfoliación (fig. 17) o la queratodermia cribiforme producida por las variantes en loricrina (fig. 18).



.")



Los autores declaran no tener ningún conflicto de intereses.



