



We present the case of a 33 year-old man with no relevant medical history who was seen for skin lesions that had been present on both pretibial areas and lateral borders of the feet for 2 years. The lesions were slightly pruritic and followed an intermittent course with periods of improvement but never healed completely. No improvement was seen with the application of topical corticosteroids. The patient reported no extracutaneous symptoms.
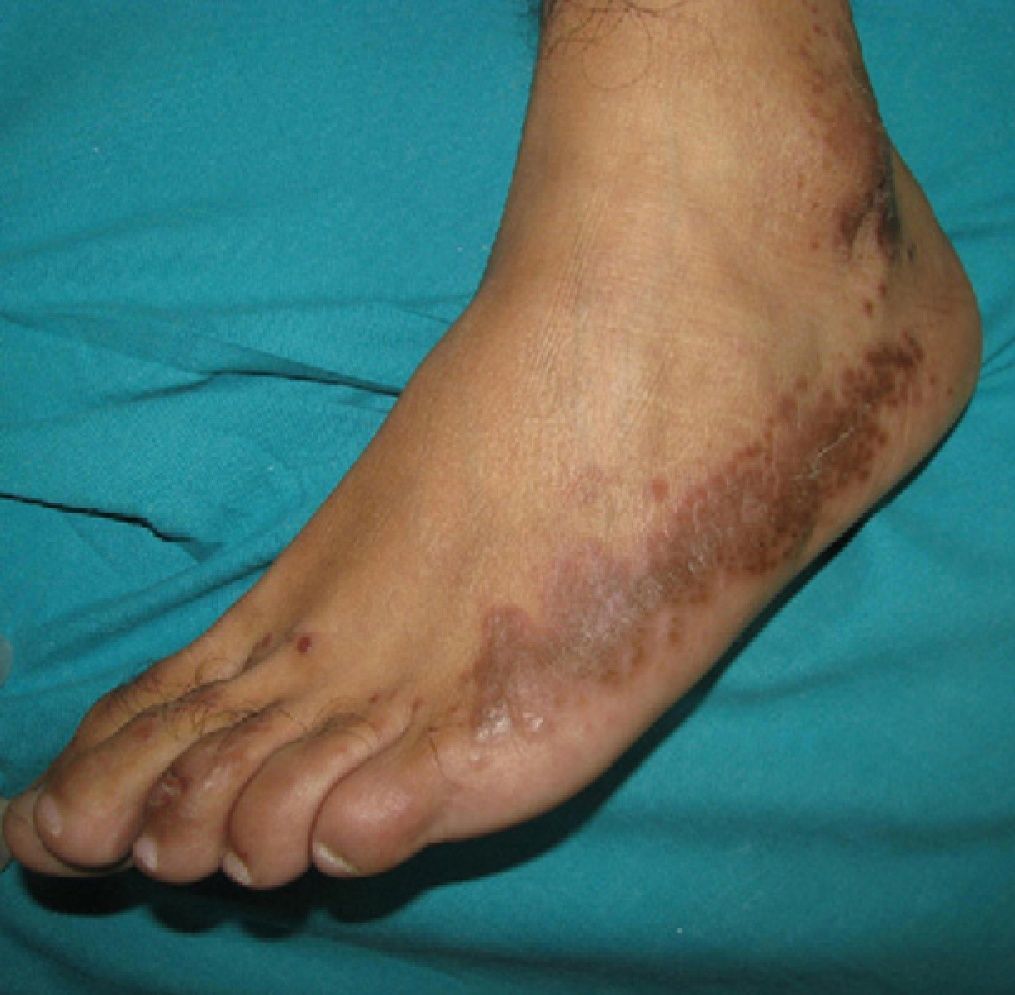
Physical ExaminationPhysical examination revealed multiple brownish papules and plaques distributed over both pretibial areas (Fig. 1). There were also multiple papules that coalesced to form brownish plaques with irregular borders on the lateral aspects of both feet (Fig. 2). These lesions extended onto the lateral surface of both ankles, where discrete ulceration was observed.
Histopathology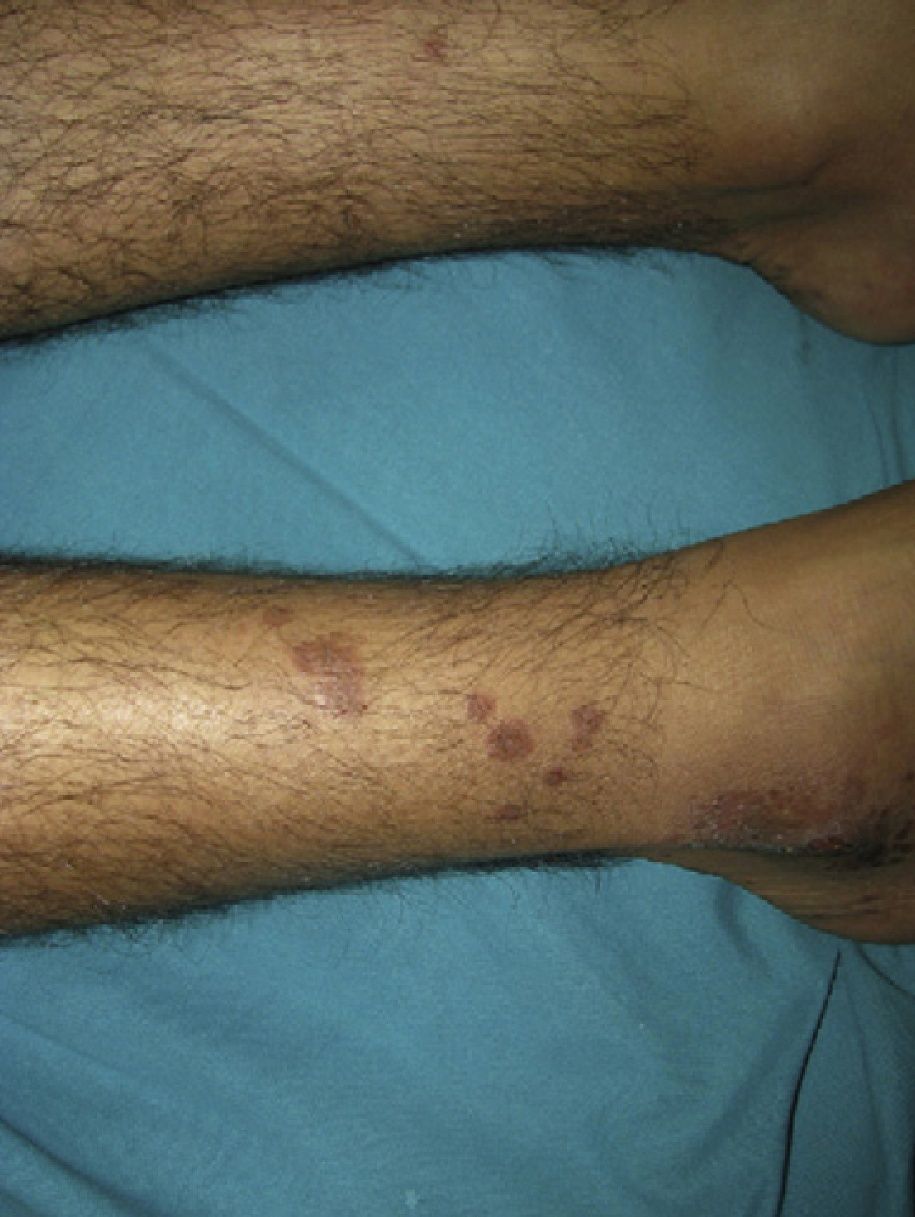
Histology showed fibrinoid necrosis of the small caliber vessels, with a polymorphonuclear infiltrate and karyorrhexis. The early stages of concentric perivascular fibrosis were also present (Fig. 3). Immunofluorescence was negative.
Additional Tests
Blood tests, liver function, kidney function, serum electrolytes, protein analysis and serum protein electrophoresis, antinuclear antibodies, and cryoglobulin were all normal. Urinary sediment was normal. Serology for human immunodeficiency virus (HIV) and hepatitis A, B, and C viruses was negative.
What Is Your Diagnosis?
DiagnosisErythema elevatum diutinum (EED).
Clinical Course and TreatmentBlood tests confirmed normal glucose-6-phosphate dehydrogenase levels, and treatment was then initiated with dapsone 100mg/d, achieving a good clinical response. After 6 weeks of treatment with no recurrence of the symptoms, the dose of dapsone was reduced to 50mg/d. At the time of writing, only a residual postinflammatory hyperpigmentation remained.
CommentEED is a rare form of leukocytoclastic vasculitis characterized by a chronic clinical course.1–3 It affects adults aged between 30 and 70 years, although cases have been reported in other age ranges. It presents clinically as erythematous-purpuric and yellowish papules that can develop into infiltrated plaques with irregular borders.2,3 The lesions are initially soft, but later acquire a firm, doughy consistency. Some patients enter a chronic phase of fibrosis in which the initial lesions are replaced by nodules that are hard and can grow to a large size.1
Histologically, EED is characterized by leukocytoclastic vasculitis of small-caliber vessels, with fibrinoid necrosis and a neutrophilic infiltrate. The epidermis shows acanthosis and parakeratosis.4 The most distinctive feature of the disease is the appearance of progressive concentric perivascular fibrosis around the affected vessels. Histological findings are very similar to those of granuloma faciale in an extrafacial location and differentiation of the 2 conditions is difficult for single lesions.5
EED has been described in association with many systemic conditions, including autoimmune and hematological diseases and infections, in particular HIV infection,6 in which EED may be the first manifestation.3 Hepatitis B or C virus infection must also be ruled out, and a complete blood count and protein electrophoresis are needed to exclude IgA and IgG gammopathy, IgA multiple myeloma, polycythemia vera, hairy cell leukemia, mixed cryoglobulinemia, and myelodysplasia.2,3 The group of diseases associated with EED includes inflammatory bowel disease, rheumatoid arthritis, systemic lupus erythematosus, and relapsing polychondritis.1,2
The treatment of choice for EED is dapsone, which reduces disease severity but is not curative; recurrence is common when treatment is stopped.2 Control of associated diseases, when present, is essential. Other therapeutic options include niacinamide, colchicine, and sulfapyridine.2 Treatment with intralesional corticosteroids and surgical excision are only indicated in cases of localized disease.2
Please cite this article as: Suárez-Pérez JA, et al. Pápulas y placas agrupadas en extremidades inferiores. Actas Dermosifiliogr.2011;102:823-824.



