



Juvenile xanthogranuloma is a non-Langerhans cell histiocytosis that typically affects children, but several cases have been reported in adults, some in connection with hematologic malignancies. We present the case of a 61-year-old woman with multiple xanthogranulomas who developed a follicular lymphoma after 4 years of follow-up. After 6 months of treatment with chemotherapy and rituximab, the cutaneous lesions disappeared and the patient achieved remission from lymphoma. We highlight this case because xanthogranuloma is a rare disorder that is difficult to diagnose in adults and also because this is the first report of an association between xanthogranuloma and follicular lymphoma. Excellent response was achieved with chemotherapy and rituximab. Finally, given the possible association between xanthogranulomas and hematologic diseases, these lesions may be a cutaneous manifestation of an occult malignancy.
El xantogranuloma juvenil (XGJ) es una histiocitosis de células no Langerhans que acontece en la edad infantil, sin embargo, se han descrito varios casos en el adulto, algunos de ellos en relación con hemopatías malignas. Presentamos el caso de una mujer de 61 años de edad con lesiones diseminadas de XGJ que a los 4 años de seguimiento desarrolló un linfoma de tipo folicular. Tras 6 meses de tratamiento con quimioterapia y rituximab se consiguió remisión del linfoma y la involución de las lesiones cutáneas. Destacamos este caso por tratarse de una entidad poco frecuente y de difícil diagnóstico en el adulto, así como por ser el primer caso asociado con linfoma folicular y que ha presentado una excelente respuesta con quimioterapia y rituximab. Además, dada su posible asociación con enfermedades hematológicas, el XGJ podría representar una manifestación de una neoplasia oculta.
Juvenile xanthogranuloma (JXG) is a non-Langerhans cell histiocytosis that typically affects young children. It is characterized by multiple yellowish-brown papulonodular lesions that resolve spontaneously. Histologically, these lesions are characterized by histiocytes and Touton-type multinucleated giant cells. Most of the few cases of JXG that have been reported in adults consist of solitary lesions that do not regress spontaneously.1 JXG has gained interest because of its possible association with oncohematologic diseases.
Case DescriptionThe patient was a 61-year-old woman (occasional smoker) diagnosed 30 years earlier with latent tuberculosis. She had undergone curettage of the uterus to treat a uterine fibroid and had required a blood transfusion. In 2004, yellowish-brown papular lesions 3 to 5mm in diameter began to appear on the patient's trunk. The lesions increased in number, gradually spreading across the abdomen and the proximal areas of the limbs (Fig. 1, A and B).

Histologic examination of one of the lesions revealed a diffuse infiltrate located mainly in the mid dermis that dissected collagen bundles and formed aggregates in the deep dermis. The infiltrate was composed primarily of cells with abundant cytoplasm and a histiocytic appearance, as well as multinucleated giant cells, some of which were Touton-type, mixed with occasional lymphocytes with no atypia and a few eosinophils (Figs. 2 and 3). Immunohistochemistry was positive for cells of monocyte-macrophage lineage (CD68) and negative for Langerhans cells (s-100 protein and CD1a). A diagnosis of adult-onset JXG was established. The patient began outpatient follow-up care and continued to develop clinically similar lesions. After 4 years of follow-up, the patient was found to have an enlarged left inguinal lymph node that was growing rapidly, with no other associated symptoms. The lymph node was excised and histology revealed a nodular lymphoproliferative process consisting of centrocytes with a germinal center phenotype (immunohistochemistry was positive for CD20, CD23, and CD10 in follicular areas, with diffuse expression of Bcl-2 and selective expression of Bcl-6 in the follicles), which indicated clonal IGH gene rearrangement (Fig. 3). The staging study revealed enlarged retroperitoneal and perivascular lymph nodes, adequate differentiation of the 3 hematopoietic cell lines, and an absence of neoplastic infiltration in the bone-marrow biopsy specimen. These findings led to a final diagnosis of well-differentiated, low-grade, stage IIIA follicular B-cell lymphoma.
.")
. B, Immunohistochemistry showing a negative result for CD1a (original magnification×200). C, Immunohistochemistry showing granular positivity for CD68 in the cellular cytoplasm (original magnification×200). D, Histology of inguinal lymph node showing a lymphoid proliferation of centrocytes (small cleaved cells) (original magnification×100).")
A, Infiltrate composed of cells with abundant cytoplasm and a histiocytic appearance, some multinucleated giant cells, lymphocytes with no atypia, and a few eosinophils (hematoxylin-eosin, original magnification×100). B, Immunohistochemistry showing a negative result for CD1a (original magnification×200). C, Immunohistochemistry showing granular positivity for CD68 in the cellular cytoplasm (original magnification×200). D, Histology of inguinal lymph node showing a lymphoid proliferation of centrocytes (small cleaved cells) (original magnification×100).
The findings of a new histologic examination of a different skin lesion, performed after the lesions had increased in number, were similar to those of the first biopsy; IGH gene rearrangement was not detected, however, due to insufficient genetic material. The patient began treatment with monthly cycles of fludarabine (40mg/d) and cyclophosphamide (250mg/d) for 3 days each month followed by a single dose of rituximab (375mg/m2); she was administered 4 cycles. She was then administered 4 cycles of a single weekly dose of rituximab (375mg/m2). Six months after completing the treatment regimen, the patient had achieved complete remission of the malignancy and the skin lesions had regressed, leaving depressed, hyperpigmented scars (Fig. 1C). After 2 years of follow-up, no new skin lesions have appeared and there is no evidence of hematologic recurrence.
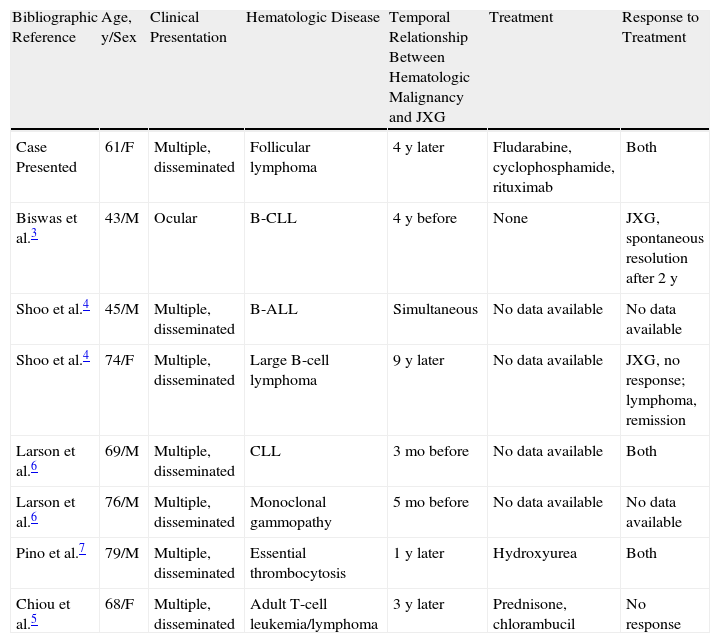
DiscussionAdult-onset JXG was first described in 1963, since which time 125 cases have been reported, most consisting of solitary lesions.1,2 Nevertheless, in the literature we have identified 21 cases with more than 10 disseminated lesions, with histologic findings similar to those seen in children with JXG.1–6 Seven of the reviewed cases of adult-onset JXG were associated with hematologic malignancies3–7 (Table 1), most frequently leukemias (3 cases),3–5 followed by lymphomas (2 cases),3,5 monoclonal gammopathy (1 case),6 and essential thrombocytosis (1 case).7 We did not find any cases of JXG associated with follicular lymphoma. The present case is therefore the first report of this association. In all cases in which JXG was associated with a blood dyscrasia, the patients presented multiple lesions with purely cutaneous involvement, with the exception of 1 case with periocular involvement.3 JXG developed before, concomitantly with, or after diagnosis of the hematologic malignancy, with intervals that ranged from 5 months to 9 years.
Cases of Adult-Onset Juvenile Xanthogranuloma Associated with Hematologic Malignancy.
| Bibliographic Reference | Age, y/Sex | Clinical Presentation | Hematologic Disease | Temporal Relationship Between Hematologic Malignancy and JXG | Treatment | Response to Treatment |
| Case Presented | 61/F | Multiple, disseminated | Follicular lymphoma | 4 y later | Fludarabine, cyclophosphamide, rituximab | Both |
| Biswas et al.3 | 43/M | Ocular | B-CLL | 4 y before | None | JXG, spontaneous resolution after 2 y |
| Shoo et al.4 | 45/M | Multiple, disseminated | B-ALL | Simultaneous | No data available | No data available |
| Shoo et al.4 | 74/F | Multiple, disseminated | Large B-cell lymphoma | 9 y later | No data available | JXG, no response; lymphoma, remission |
| Larson et al.6 | 69/M | Multiple, disseminated | CLL | 3 mo before | No data available | Both |
| Larson et al.6 | 76/M | Multiple, disseminated | Monoclonal gammopathy | 5 mo before | No data available | No data available |
| Pino et al.7 | 79/M | Multiple, disseminated | Essential thrombocytosis | 1 y later | Hydroxyurea | Both |
| Chiou et al.5 | 68/F | Multiple, disseminated | Adult T-cell leukemia/lymphoma | 3 y later | Prednisone, chlorambucil | No response |
Source: Adapted from Shoo et al.4 Abbreviations: B-ALL, B-cell acute lymphoblastic leukemia; B-CLL, B-cell chronic lymphocytic leukemia; F, female; JXG, juvenile xanthogranuloma; M, male.
The response of the JXG and the tumor to treatment targeting the hematologic neoplasm was variable. Remission of the tumor and regression of the skin lesions occurred in only 2 cases.6,7
Some studies have reported that children who have JXG and neurofibromatosis have an increased risk of developing leukemia, but doubts about this association have subsequently been raised.3 Two cases have been reported in children of JXG with disseminated cutaneous involvement associated with myeloproliferative diseases. One report described a 3-year-old boy who was diagnosed with JXG and 6 months later developed B-cell acute lymphoblastic leukemia (B-ALL).8 The patient failed to achieve remission with chemotherapy, although the skin lesions did flatten slightly. The other case was a newborn with JXG and hemophagocytic lymphohistiocytosis who developed chronic juvenile myelomonocytic leukemia.9 Castro et al.10 described the cases of 3 children between the ages of 8 and 13 years who presented JXG lesions in unusual locations—lymph nodes, bones, and lungs—in association with ALL. The intervals between the appearance of JXG and the onset of ALL ranged from 1 to 12 years. However, given the higher incidence of this disease in children, its self-resolving course, and reports of atypical forms of presentation, we believe that the association between JXG and blood neoplasms is weaker in children.
Regarding the etiology and pathogenesis, it has been speculated that histiocytoses develop as a result of the proliferation of a common CD34+ precursor cell in the bone marrow. The abnormal multiplication of these cells is caused by the action of various stimuli, including cytokines or gamma globulins produced by an underlying malignancy.11 Moreover, it has been suggested that some cases of Langerhans cell histiocytosis associated with myeloproliferative or lymphoproliferative diseases may have a common origin in a hematopoietic precursor cell capable of dedifferentiating both into tumor cells, which leads to the neoplasm, and into cells of monocyte-dendritic lineage, which leads to histiocytosis.12 A recent study described a case of JXG with visceral involvement (liver and spleen) in a child aged 5 years.13 Three months after the patient was diagnosed with T-ALL, clonal biallelic rearrangement of TCRγ in blasts and histiocytes was detected. In our patient, we were unable to establish a clonal relationship between the lymphoproliferative cells and the histiocytes that formed part of the skin lesions because the skin biopsy specimen contained insufficient genetic material. In the literature, we found no other cases of JXG with clonal rearrangement among either the hematologic tumor cell population or the histiocytosis. It would therefore be interesting, in future studies, to demonstrate this association in patients with both diseases. This research could be extended to other cases of blood dyscrasias associated with various types of non-Langerhans cell histiocytosis, such as xanthoma disseminatum and necrobiotic xanthogranuloma.
No treatment regimen has been shown to cure disseminated adult-onset JXG. Patients with JXG and neoplastic disease whose skin lesions improved did so in response to antineoplastic therapy. Our patient received chemotherapy with fludarabine and cyclophosphamide in combination with rituximab (an anti-CD20 monoclonal antibody that inhibits CD20+ lymphocytes). The cytostatic treatment induced apoptosis in the neoplastic cells, thereby inhibiting the production of various cytokines and other mediators that might otherwise have participated in histiocytic proliferation. Various mechanisms by which rituximab may inhibit lymphocytes have been proposed, including modulation through the macrophage-histiocyte system,14 which could have contributed to the resolution of JXG in our patient. Our review of the literature has revealed no previous reports of disseminated JXG being treated with rituximab, although there has been a description of complete resolution of a necrobiotic xanthogranuloma associated with lymphocytic lymphoma in a patient who followed the same treatment regimen as our patient.15
ConclusionThis is the first report in an adult of disseminated JXG associated with follicular lymphoma with an excellent response to cytostatic treatment and rituximab. This result supports the hypothesis that JXG is associated with hematologic malignancies. Not enough cases have been reported for this entity to be considered a true paraneoplastic syndrome. Nevertheless, it is advisable to follow up these patients using general physical examination and additional tests to screen for tumors—especially hematologic ones—in those patients who have numerous lesions that have appeared progressively.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Narváez-Moreno B, et al. Xantogranuloma juvenil diseminado del adulto asociado a lin-foma folicular, respuesta completa a quimioterapia y rituximab. Revisión de la literatura. Actas Dermosifiliogr. 2013;104:242–6



