








Vascular occlusion has multiple, diverse clinical manifestations, some of which can have grave consequences for patients. It also has a wide variety of causes, including thrombi, which we recently addressed in part I of this review. In this second part, we look at additional causes of vascular occlusion.
La patología vascular oclusiva es causante de diversas y variadas manifestaciones clínicas, algunas de las cuales son de catastróficas consecuencias para el paciente. Dado que las causas de tal oclusión son muy variadas, hemos abordado en un artículo previo reciente en esta misma revista, las causas trombóticas. En el presente artículo, recopilamos diversas causas adicionales de oclusión intravascular.
The previous part of this 2-part review discussed the main causes of thrombotic vascular occlusion. This second part addresses a diverse collection of causes of vascular occlusion. The clinical signs of many of the occlusive events discussed are similar, however. They include diverse signs of ischemia, notably livedo reticularis, ulcers and more. Histopathology to identify the underlying cause of occlusion therefore plays a key role.
Coagulation DisordersSystemic Coagulation DisordersThis category includes clinical pictures that develop with systemic coagulation deficiencies generally due to changes in the levels of factors and proteins involved in the process.
Protein C and S deficienciesDefinitionThe protein C and S system is an important inhibitor of coagulation in small vessels in particular. Deficiencies in the system can give rise to severe microvascular occlusion syndromes that lead to purpura fulminans.1,2
Clinical presentationSigns vary according to the degree of the deficiency. Homozygous mutations that seriously disrupt the system can cause severe symptoms of neonatal purpura fulminans, with retiform purpura and gangrene on the extremities.1,3 Partial deficiencies, on the other hand, in heterozygous mutations might pass unnoticed or manifest only in the presence of some external trigger, such as treatment with oral anticoagulants.1,2
HistopathologyHistopathologic findings are similar to those in other noninflammatory microvascular occlusive syndromes. Soft thrombosis of vessels of varying sizes is observed in the dermis and hypodermis. An inflammatory infiltrate is scant or nil, without signs of vasculitis.
Warfarin- or coumarin-induced necrosisDefinitionAcenocoumarol and warfarin are widely used oral anticoagulants that work by inhibiting vitamin K, reducing plasma levels of vitamin K-dependent coagulation factors II, VII, IX, and X. These anticoagulants also affect the protein C and S inhibitor system.4 Because these proteins have short half-lives, a paradoxical procoagulant effect is observed in the early days of treatment.5,6 These anticoagulants have also recently been linked to nonuremic calciphylaxis.7
Clinical presentationCutaneous necrosis associated with anti-vitamin K therapy typically presents during the first week of treatment, coinciding with a relative decrease in protein C and protein S levels.4,5 Necrosis characteristically develops in areas where adipose tissue tends to accumulate, such as the breasts and thighs. Signs and symptoms overlap with those of other disorders that manifest with retiform purpura and erythematous and hemorrhagic lesions with characteristic necrotic centers; there may be branching features in the borders of lesions.4,7 Warfarin-associated nonuremic calciphylaxis also presents with retiform purpura, but that sign appears after months or years of treatment.7
HistopathologyBiopsies of classic cutaneous necroses due to vitamin K antagonists typically show signs of noninflammatory thrombotic vasculopathy4,5 with eosinophilic fibrin-like deposits occluding multiple vessels.4 Affected vessels vary in size; they may be superficial ones in the papillary dermis, or deeper in the reticular dermis, or still deeper at the dermal-hypodermal junction.4–6 It is common to observe extravasated blood cells surrounding the occluded vessels as well as associated epidermal necrosis.4 A very scant inflammatory infiltrate may be present, though there may be none at all; vasculitis is not present.4,5 The findings in warfarin-induced nonuremic calciphylaxis overlap with those of classic calciphylaxis.7
Disseminated intravascular coagulationDefinitionDisseminated intravascular coagulation (DIC) is an acquired syndrome triggered by various factors. When coagulation is activated, fibrin is deposited in vessels, obstructing them and compromising the blood supply to organs and systems. Coagulation proteins and platelets are consumed, leading to significant bleeding. A diagnosis of DIC requires the presence of an underlying disease. Certain analytical criteria must be met. The most common cause is bacterial sepsis.8,9
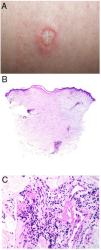
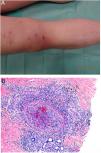
Clinical presentationDIC involves multiple organs. The first sign often takes the form of skin lesions, typically a petechial rash and purpuric plaques that may progress to hemorrhagic blisters. Two common conditions that often develop in the context of DIC are purpura fulminans and symmetrical acral gangrene; the disease does not always progress to that degree, however. Purpura fulminans is characterized by confluent purpuric lesions that progress rapidly to well-circumcised necrosis. Mortality can reach 50%. Symmetrical acral gangrene consists of symmetrical ischemia in distal areas10,11 (Fig. 1A).
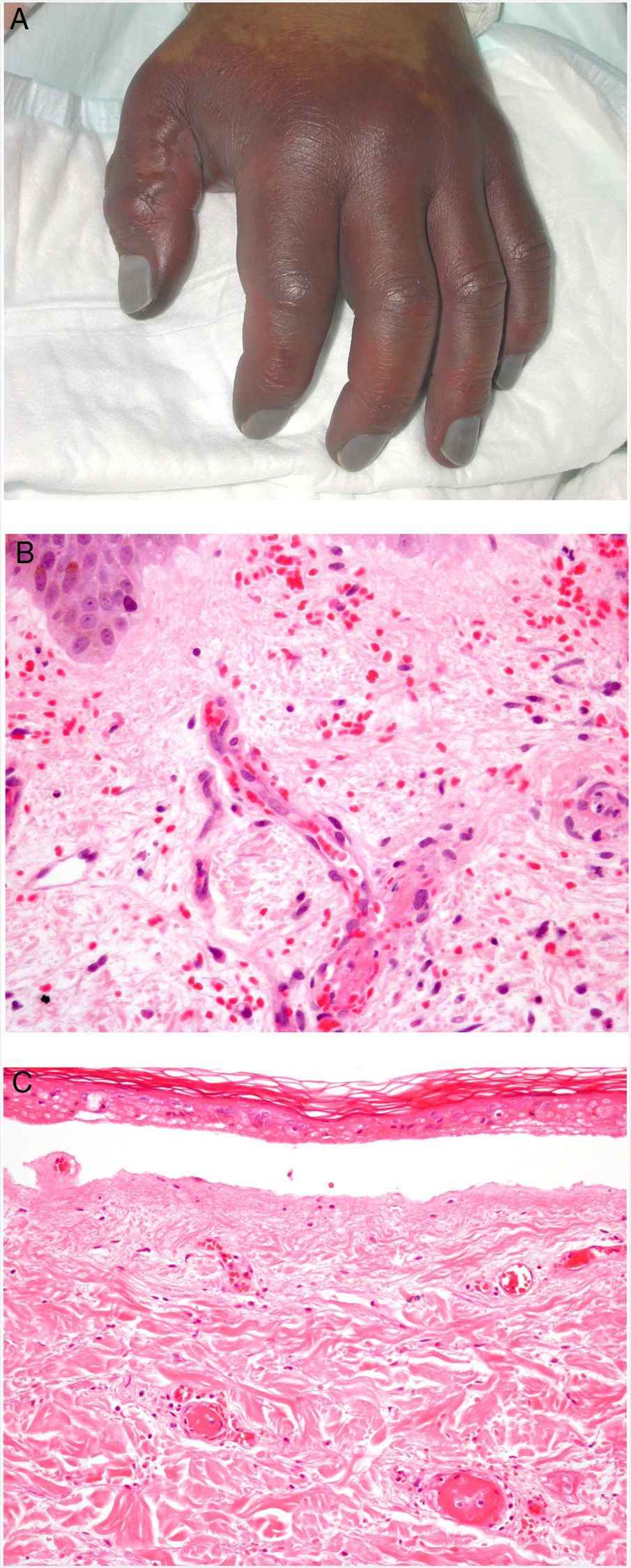
A, Symmetrical acral gangrene, characterized by a well-circumscribed ischemic plaque on the left hand. Note the bluish discoloration of the nails. B, Fibrin thrombi in the superficial vascular plexus. Note the extravasation of red blood cells in the dermis. Hematoxylin-eosin, magnification × 400. C, Disseminated intravascular coagulation a few days old. Note the formation of a subepidermal blister, epidermal necrosis, and fibrin thrombi in small dermal vessels. Hematoxylin-eosin, magnification × 200.
Histopathologic findings in DIC are not pathognomonic but may point to diagnosis in the context of certain clinical pictures. Thrombi containing fibrin and platelets are found, mainly in small dermal blood vessels. They vary in number from few to many. Occlusion of small arteries in the reticular dermis is seen less often. Red blood cell extravasation is also common, especially so in cases of purpura fulminans (Fig. 1B). An inflammatory infiltrate is usually scant or nonexistent. Biopsies of lesions only a few days old may display epidermal necrosis, subepidermal blistering, and necrosis of eccrine glands and hair follicles (Fig. 1C). Lesions that have progressed to gangrene show extensive necrosis in all skin structures.10 Bearing in mind that sepsis is the most common cause of DIC, mixed patterns of thrombotic and septic vasculopathy with fibrinoid necrosis or bacterial invasion of the vessel wall are often observed. An inflammatory infiltrate is more prominent in such cases.12
Antiphospholipid syndromeDefinitionAntiphospholipid syndrome is an arterial or venous thrombotic autoimmune disease associated with the presence of antiphospholipids (lupus anticoagulant, anticardiolipin antibody, and anti-β2 glycoprotein). This syndrome is probably the most common acquired thrombophilic disorder. It may be idiopathic or associated with various other conditions, among them autoimmune diseases (especially lupus erythematosus), infections, hematologic or solid tumors, and other less frequent scenarios.13
Clinical presentationPatients are most often young women. The early signs are usually skin necrosis, livedo reticularis, Raynaud phenomenon or subungual bleeding, particularly in primary forms associated with lupus. Diagnosis requires a relevant clinical sign along with a finding of one of the antibodies listed above.14 The relevant clinical criteria are confirmed vascular thrombosis in any location (skin, central nervous system, heart, lung, kidney, digestive tract) or a history of gestational problems (intrauterine fetal death, spontaneous abortion, premature delivery). Patients should be followed for 5 to 10 years after diagnosis because many develop systemic lupus erythematosus. Catastrophic illness develops in fewer than 1% of patients with antiphospholipid syndrome, but mortality exceeds 50% in such cases; death is usually due to generalized thrombotic microangiopathy involving multiple organs simultaneously or over a short period of time.15
HistopathologyLook for fibrin thrombi in the absence of inflammation of the vessel wall, in arteries or veins of any caliber (Fig. 2A–B). A few lymphocytes or plasma cells may occasionally be observed. Neovascularization and vascular proliferation (reactive angioendotheliomatosis) may occur when disease is of long duration.
Vascular Coagulopathy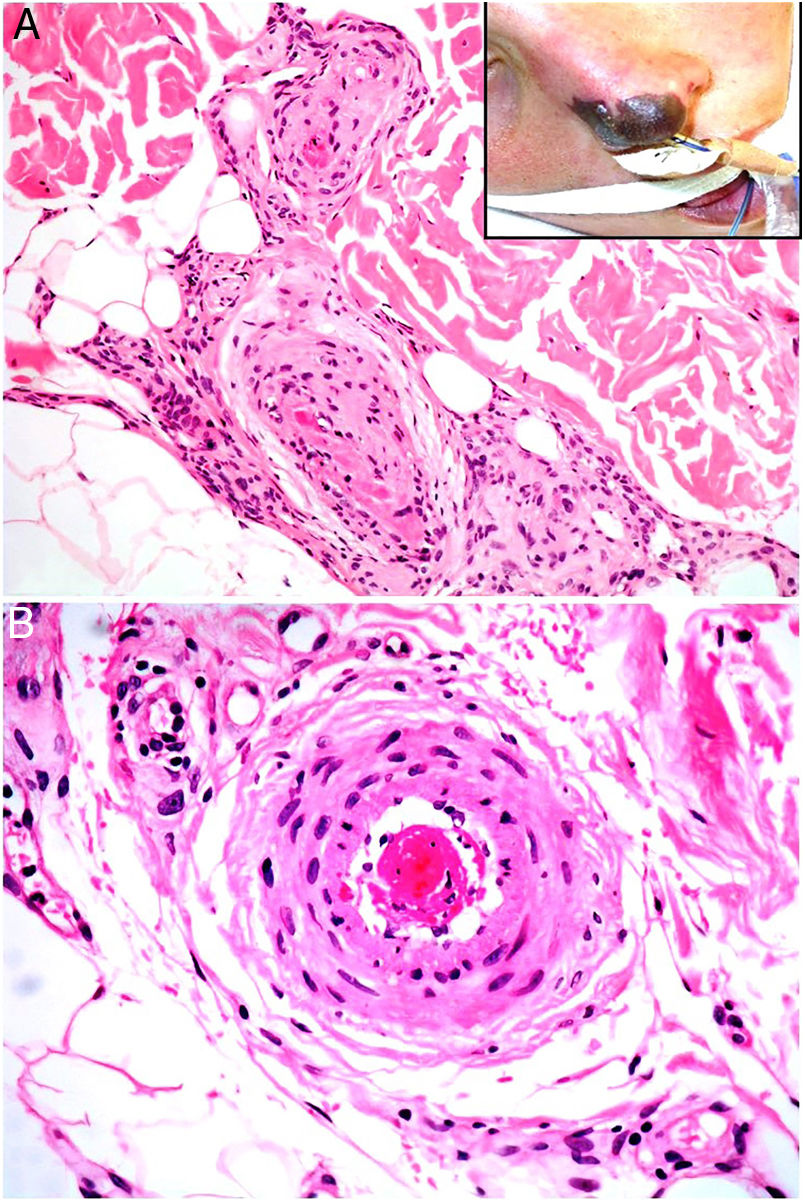
Defective coagulation may be triggered by abnormalities in the structure or composition of vessel walls. Generally the defects are in the endothelium.
Sneddon syndromeDefinitionSneddon syndrome is a rare, episodic or chronic neurocutaneous syndrome that progresses slowly and is characterized by generalized livedo racemosa and repeated strokes.16 More than 50% of patients have the primary form. The secondary form occurs in association with an autoimmune disease or a thrombophilic disorder (antiphospholipid syndrome). A familial form involving a mutation in the CECR1 (cat eye syndrome region, candidate 1) gene has been described; this form produces adenosine deaminase 2 deficiency.17
Clinical presentationAround 80% of patients are women; the mean age at diagnosis is 40 years, although onset can also occur in childhood. The main skin sign is livedo racemosa, although the characteristic lesions were at first described as livedo reticularis.18 They usually appear first on the lower back and progress to the back of the arms and thighs, sparing the feet and face. The lesions are not painful, edema and ulceration are rare. The severity of skin signs does not correlate with neurologic symptoms. Rarely, acral cyanosis, Raynaud phenomenon, angiomatosis, circumscribed skin ulcers, atrophic annular lichen planus, or gangrene may be observed. Neurologic manifestations tend to begin with headache, nausea, and dizziness. Recurrent transient ischemic attacks and finally ischemic or hemorrhagic strokes ensue. Progressive cognitive decline and dementia occur in this phase.
HistopathologyDeep biopsy samples from the center, not the ring, of livedo racemosa lesions offer the best diagnostic yield in histologic evaluation.16 The earliest lesions, which are rarely biopsied, are endothelial and show inflammation in the walls of small- and medium caliber arteries. The most common observation is subendothelial thickening due to myocyte proliferation leading to total or subtotal occlusion of dermal and hypodermal blood vessels. Local hypoxia stimulates endothelial proliferation and adventitial angiogenesis in the final phase.19
Livedoid vasculopathyDefinitionLivedoid vasculopathy is a common chronic thrombotic skin disease characterized by ischemia. The main causes are coagulation disorders, autoimmune diseases, and certain infections. Venous insufficiency, a very important facilitator of thrombosis, can be added to the list.
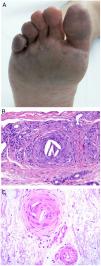
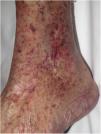
Clinical presentationThis diagnosis is seen more often in middle-aged women, who develop papules and plaques on the lower extremities. The lesions are more or less purpuric, symmetrical, painful, and persistent. They give way to slowly progressing ulcers that, depending on size, may heal or lead to porcelain-white scars known as atrophie blanche. Thrombosis is caused by a hypercoagulability state due to circulating procoagulant antibodies (ie, cryoglobulins, antibodies in idiopathic antiphospholipid syndrome, or those appearing in autoimmune diseases such as lupus erythematosus, viral hepatitis, etc). Thrombocytosis might also be the culprit, or there may be hereditary abnormalities affecting coagulation factors (eg, Factor V Leiden mutation or fibrinolytic factor deficiency states). Chronic venous insufficiency and injuries must be added to the conditions that facilitate thrombosis. The condition is idiopathic in many cases, however. Skin signs are purpuric papules and plaques that tend to ulceration, leading to porcelain-white scars (Fig. 3A).
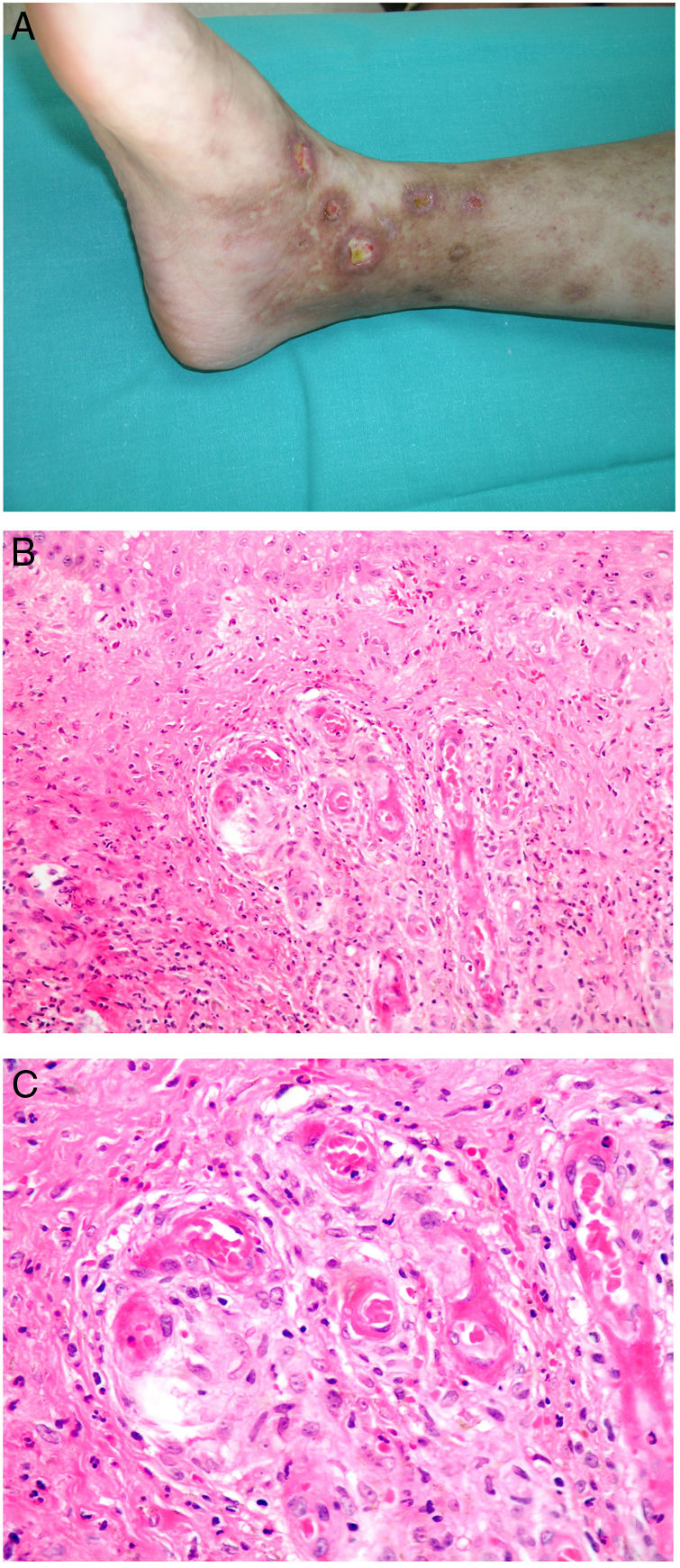
Livedoid vasculopathy. A, Erythema, multiple ulcers, and discoloration of the skin due to red cell extravasation on the leg of a 62-year-old woman with lupus erythematosus and venous insufficiency. B, Glomerular-like vascular aggregates in the superficial dermis. Hematoxylin-eosin, magnification × 100. C, Detail of the previous microphotograph. Note the vascular wall thickening, elevated number of pericytes, and homogeneous deposits of eosinophilic fibrinoid material. Around the blood vessel, note extravasated red cells, occasional hemosiderin-laden macrophages, a lymphomacrophagic infiltrate, and collagen fibrosis; Hematoxylin-eosin, magnification × 200.
The skin biopsy should include tissue adjacent to the ulcerated area to demonstrate the condition of the surrounding skin. There are no pathognomonic histologic signs. Livedoid vasculopathy affects vessels in the superficial to mid-dermis. Present are glomerular-like aggregates of dilated vessels, periodic acid–Schiff (PAS)-positive fibrinoid hyaline deposition in the walls, thrombosis, and bleeding that should not be confused with vasculitis. A variable perivascular lymphomacrophagic infiltrate can also be observed. Ulcers give way to scarring. Epidermal atrophy and intense collagen fibrosis due to chronic ischemia appear in the most advanced cases. This development is the cause of the characteristic clinical sign of porcelain-white atrophy (Fig. 3B and C). Direct immunofluorescence can reveal deposits of immunoglobulins, complement, and fibrinogen, suggesting the nature of the immune process. Other thrombosing vascular diseases must be ruled out in the differential diagnosis, for which reason clinicopathologic correlation is important.
Livedoid vasculopathy is histologically distinguishable from type I cryoglobulinemia and diabetic microangiopathy, neither of which show a glomerular-like vascular architecture. Distinguishing this diagnosis from stasis dermatitis or acroangiodermatitis is complicated, but these last 2 entities generally show marked ferrous deposition or intense vascular proliferation, respectively.
Degos diseaseDefinitionDegos disease, also called Kohlmeier–Degos disease or atrophic papulosis, is rare: fewer than 200 cases have been reported.20 Patients are mainly women between the ages of 20 and 50 years, although cases have also been reported in children.21 The etiopathogenesis is unknown, but associated coagulopathy and vasculitis of the endothelial lining, possibly related to complement factors, have been reported.20–22 Reports of familial cases suggest the possibility of genetic predisposition.
Clinical presentationThe following lesions are considered pathognomonic: atrophic papules measuring less than 1 cm, with porcelain-white centers and telangiectatic erythematous halos21 (Fig. 4A). A characteristic “crown of thorns” pattern is visible under a dermoscope.23 The trunk and extremities are affected but not the scalp or face. Two forms of Degos disease are currently considered. A benign form, affecting mainly the skin, has a good prognosis and accounts for 71% of the cases. The second form is systemic and involves malignant atrophic papulosis. It may be concurrent with the cutaneous form or develop later. The systemic form can lead to serious complications and even death in 75% of the cases due to cerebral, lung, kidney or other organ involvement; intestinal perforation may occur.20,21
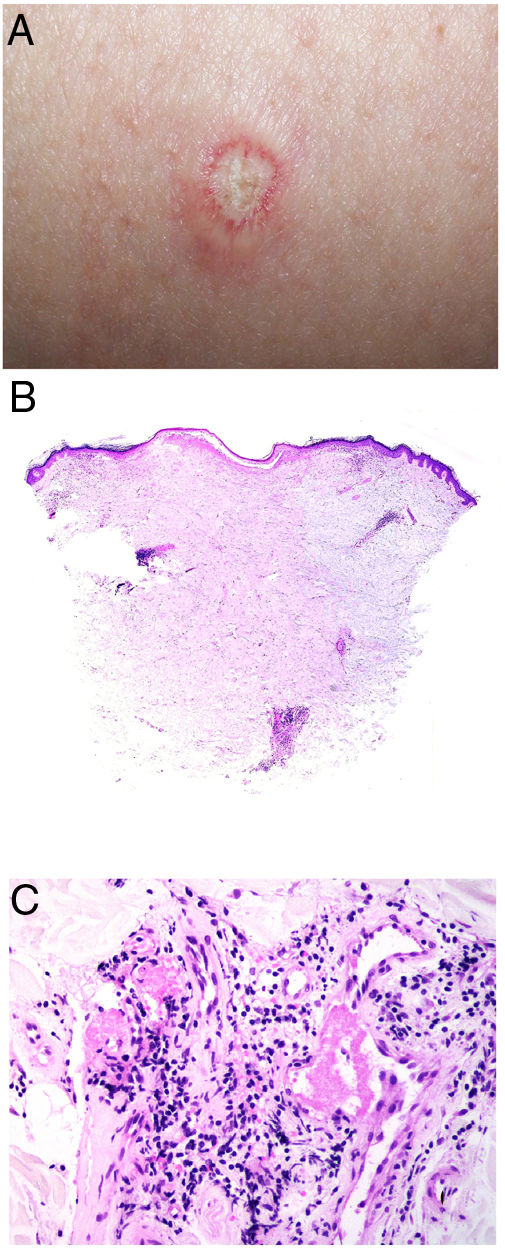
Degos disease. A, Atrophic papule with a porcelain-white center and a telangiectatic, erythematous halo. This lesion is considered a pathognomonic finding in this disease. B, Wedge-shaped infarct with a vertex extending downward. At the point of the vertex, a slight, chronic, perivascular lymphocytic infiltrate can be seen around the vessel. Hematoxylin-eosin, magnification × 20. C, Detail of the vessel shown above. Note the numerous intravascular thrombi. Hematoxylin-eosin, magnification × 400.
Findings depend on when in the course of disease the examination takes place. Characteristic signs of established disease are vascular lesions and wedge-shaped infarcts in the dermis (Fig. 4B). Endothelial swelling and intraluminal fibrin thrombi can be observed (Fig. 4C). The nearby area of the dermis contains perivascular and periadnexal lymphocytic inflammatory aggregates and interstitial mucin deposition. The epidermis might show ischemic damage, lesions at the interface, and hyperkeratosis.24 Early lesions can be similar to cutaneous lupus erythematosus, with perivascular lymphocytic aggregates and interstitial mucin deposition.25
Occlusive Syndromes Involving Red Blood CellsIn some clinical pictures, aggregates of red cells are the main culprits in vascular occlusion.
Stress reticulocyte adhesionDefinitionStress reticulocyte adhesion is caused by changes in the rheologic properties of red cells, favoring viscosity and the propensity to thrombosis.26 Although various diseases can alter red cell rheology, this concept is applied mainly in sickle-cell anemia, in which an intraluminal occlusion is due to a 2-step mechanism. CD36+ stress reticulocytes that are released tend to adhere to the endothelium, increasing the expression of ICAM-1 (intercellular adhesion molecule 1). These processes activate coagulation, and the resulting hypoxia increases the release of new stress reticulocytes, with further expression of ICAM-1.
Clinical presentationPainful single or multiple ulcers develop on the skin, especially on the internal surface of the ankle.27 Stroke, pulmonary hypertension, splenic infarction, and changes in kidney function secondary to thrombotic events in various organs can also occur.
HistopathologyEpidermal ulcers with granulation tissue are observed. Intraluminal fibrin thrombi may form, and vessel walls may show hyalinization, as in livedoid vasculopathy.28 Acanthosis and keratosis can usually be seen in the epidermis adjacent to the ulcer.
EmboliAn embolism is any structure carried in circulating blood that partially or totally obstructs a vessel. An embolism may be a thrombus that detaches and is then carried to another site. However, it may also be formed of materials present in the blood stream but not themselves blood components. They may even be totally foreign to the human body.
Endogenous MaterialCholesterolDefinitionA multisystemic disorder releases cholesterol crystals from atherosclerotic plaques into the blood stream, which carries them distally into medium- and small-caliber arteries, where they partially or completely occlude the lumen. Arteries in skin can be affected. Although crystals may be released from a plaque spontaneously, the event is generally preceded by an angioinvasive procedure or the use of destabilizing anticoagulants or fibrinolytic agents.29
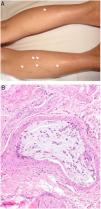
Clinical presentationSigns are highly varied, but the most common ones are livedo reticularis, purpuric papules and nodules, petechiae, and cyanosis affecting distal areas of the extremities (Fig. 5A). Ulcers and gangrene may appear in the most severe cases.
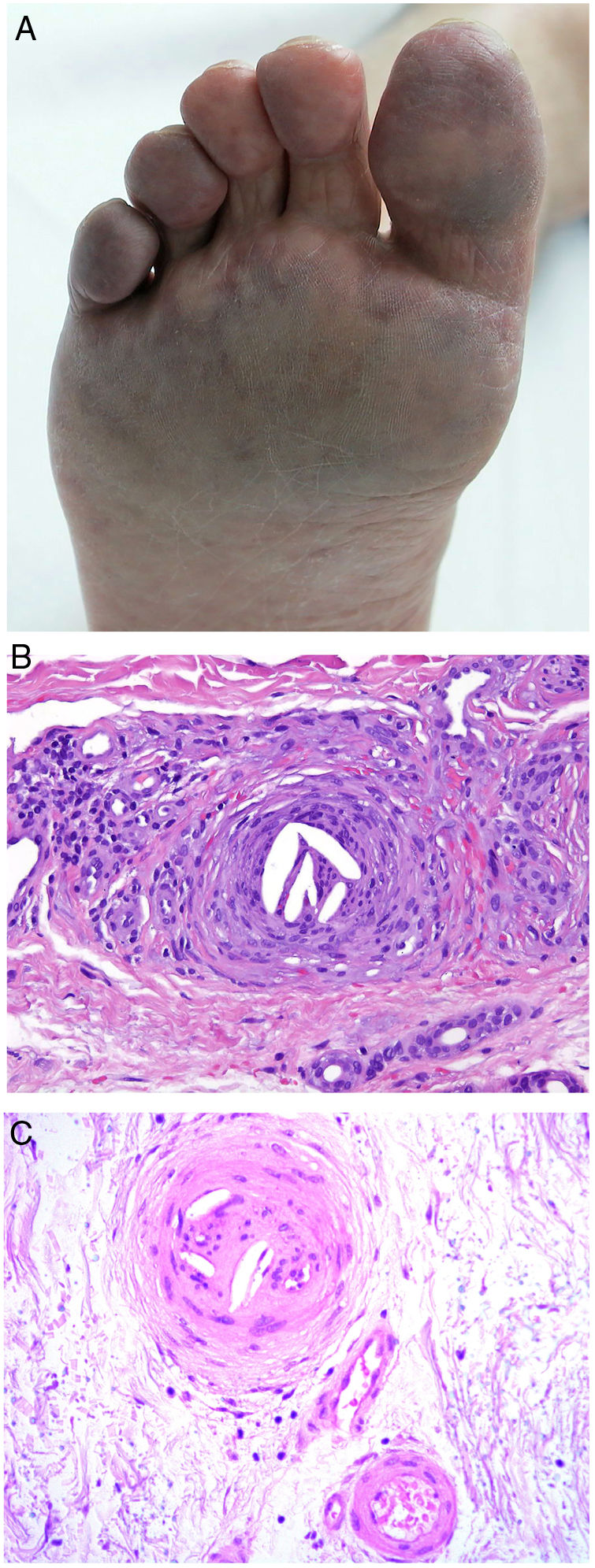
A, Livedo reticularis on the sole of the right foot of a patient with cholesterol crystal emboli. B, Cholesterol crystal emboli. Empty acicular spaces inside a blood vessel corresponding to cholesterol crystals that were dissolved during preparation of the biopsied tissue. A tissue response can be observed in the form of giant multinucleated cells surrounding the empty acicular spaces. Hematoxylin-eosin, magnification × 200. C, Late-phase emboli may show fibrosis surrounding the crystals and complete occlusion of the blood vessel. Hematoxylin-eosin, magnification × 200.
A highly characteristic, specific finding is the presence of needle-shaped, biconvex clefts inside at least 1 small- or medium caliber vessel (100–200 μm) (Fig. 5B). These shapes are the negative image of cholesterol crystals dissolved while the biopsy specimen was being prepared. They are usually found in vasculature at the dermal-hypodermal junction.30 However, it may be necessary to prepare several sections in order to locate these crystals. Other common findings are neutrophils and eosinophils surrounding the crystals in early lesions and multinucleated cells and fibrosis in late phases of disease16 (Fig. 5C).
OxalateDefinitionOxalate deposition mainly occurs in primary hyperoxaluric states, a group of inherited autosomal recessive metabolic disorders in which serum oxalate is overproduced and subsequently deposited in tissues. Secondary oxalosis is the result of excessive intake or supply of oxalate or its precursors (ethylene glycol poisoning, the anesthetic methoxyflurane, ascorbic acid). Various intestinal diseases, ileal resection, and chronic hemodialysis may also lead to an oversupply of oxalate.
Clinical presentationSymptoms typically occur at an early age and mainly affect the kidneys (recurrent urinary calculi, and renal failure). Intravascular oxalate deposits in the skin cause livedo reticularis, acral cyanosis, peripheral gangrene, and ulcers.31
HistopathologyMedium-caliber vessels are occluded by brownish crystalline material. The birefringent, crystals are acicular, possibly rectangular, and are visible in polarized light. Findings may include dermal and hypodermal necrosis.32
Crystal globulin vasculopathy: crystalglobulinemiaDefinitionCrystalglobulinemia consists of the irreversible crystallization of monoclonal proteins inside the blood vessels of patients with multiple myelomas or gammopathy of uncertain relevance.33 Fewer than 30 cases have been reported in the literature, but this entity is probably not as rare as is believed. When the monoclonal protein crystalizes on exposure to cold, the disease is referred to as cryocrystalglobulinemia. The precipitated crystals cause endothelial damage, activating the coagulation cascade, thrombosis, occlusive events, and subsequent ischemic damage.
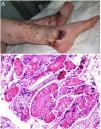
Clinical presentationSkin ulcers and purpuric lesions are usually seen on distal portions of extremities (Fig. 6A). If the kidney is involved, renal function may be severely impaired, with bilateral renal artery thrombosis. Other organs may also be affected, causing such complications as peripheral neuropathy and polyarthropathy.34
Histopathology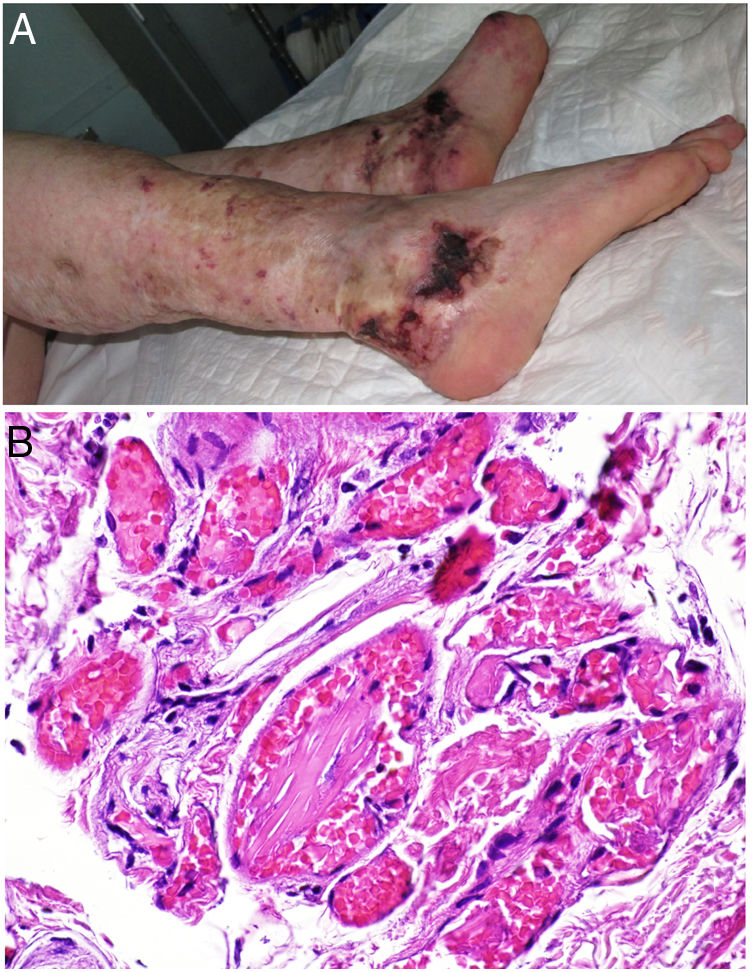
Thrombotic vasculopathy caused by intraluminal crystalloid structures can be observed in small- and medium caliber blood vessels (Fig. 6B). The PAS-positive, nonbirefringent crystals are visible in polarized light and surrounded by fibrin and red cells. Immunofluorescence demonstrates the component monoclonal immunoglobulins. Crystal morphology varies: they may be needle-shaped, rhomboidal, rectangular, or cuboid. There is no correspondence between the shape of crystals and the type of immunoglobulin forming the embolus or clinical signs.35,36
TumorsAtrial myxomaDefinitionWhile rare, myxomas are the most frequent cardiac tumors. The left atrium is affected in 75% of cases. Patients are usually women between the ages of 30 and 60 years.37 Ninety percent of cases are sporadic. The rest occur in the context of Carney complex or other hereditary diseases linked to mutation in the protein kinase CAMP-dependent type I regulatory subunit alpha (PRKAR1A) gene.16 A cardiac myxoma is not often suspected because constitutional symptoms are mild and the usual test battery gives nonspecific results. Fragments from these myxomas cause emboli in 30% to 40% of cases, preferentially in the central nervous system and skin.37–39
Clinical presentationMyxomas typically lead to recurrent episodes of transient, usually painful, skin lesions on the extremities in the form of erythematous macules or papules that are more or less purpuric (Fig. 7A). Other reported skin changes include livedo reticularis, digital cyanosis, petechiae, acral necrosis or ulcers, subungual splinter bleeding, annular lesions or a serpiginous eruption, and swollen hands or feet.16,37–40 A skin biopsy can be key, given that cutaneous emboli are sometimes the first or only manifestation of disease. Biopsies must harvest tissue from the deep dermis or the hypodermis, given that changes usually occur within arterioles.
Histopathology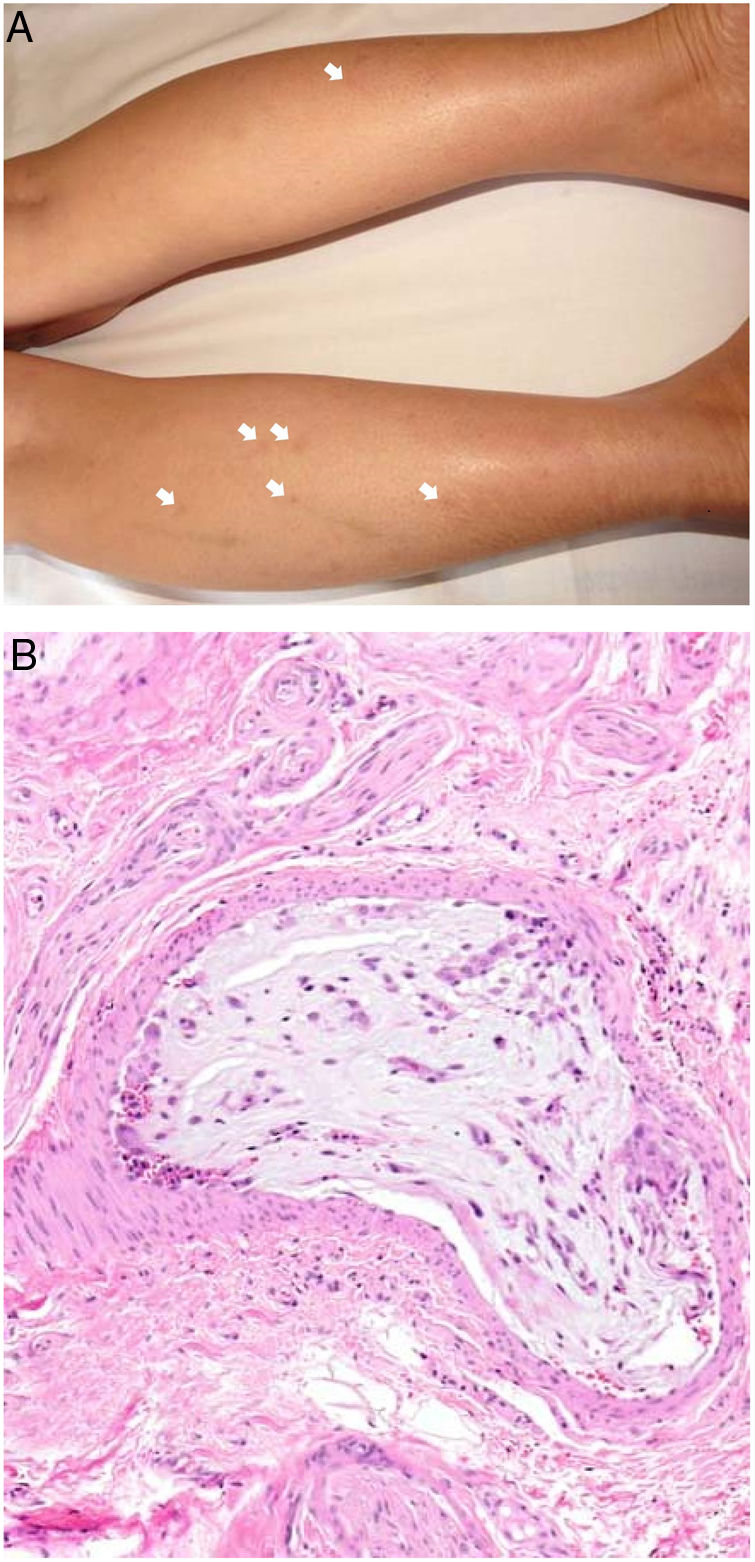
Biopsied tissue is often normal or shows nonspecific changes such as intravascular fibrin deposition. Only exceptionally does an arteriolar lumen appear occluded by lax, highly vascularized myxoid material that contains stellate or spindle cells (Fig. 7B). Lesions are stained with colloid iron and Alcian blue.16,37–39
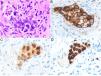
Intravascular large B-cell lymphomaDefinitionIntravascular large B-cell lymphoma (IVLBCL) is a rare entity defined by a proliferation of neoplastic large B cells mainly or exclusively inside blood vessels, especially capillaries.41
Clinical presentationThe average age of patients at the time of diagnosis is 70 years, and the clinical presentation is heterogeneous. Three clinical variants have been described. The first is the classic form, characterized by fever and other “B” symptoms, compromised functional status, skin lesions, altered level of consciousness, and hypoxemia for no clear reason. The second is the cutaneous variant, which presents with maculopapular eruptions, palpable purpura, orange-tinged skin, cellulitis, purplish erythematous nodules (some of which progress to ulcers) in the absence of systemic compromise. The third is associated with hemophagocytic syndrome.42,43
HistopathologyCell morphology varies in IVLBCL. Centroblasts, immunoblasts, and plasmablasts are observed. Their intravascular growth pattern may be dyscohesive (isolated intraluminal cells), cohesive (cells occluding the lumen), or marginal (cells adhering to the endothelium). The atypical cells are CD20+ and for the most part do not derive from germinal center B cells (CD10−, BCL6+/−); they express MUM-1 proteins (Fig. 8). Coexpression of CD5 has been reported in nearly 40% of cases, and the Ki-67 proliferation index is usually elevated (around 90%).44,45 The role of nonlesional skin biopsy for the diagnosis of IVLBCL is an important issue currently under debate. Even though there is no consensus on its utility in the literature, biopsies that sample adipose tissue have been reported to offer good sensitivity (near 80%) and specificity near 100%.46 Atypical CD30+ T-cell proliferation found in tissue samples requires particular attention in the differential diagnosis of IVLBCL. This type of pseudolymphoma has been described in relation to inflammatory diseases, tumors, and trauma.47,48
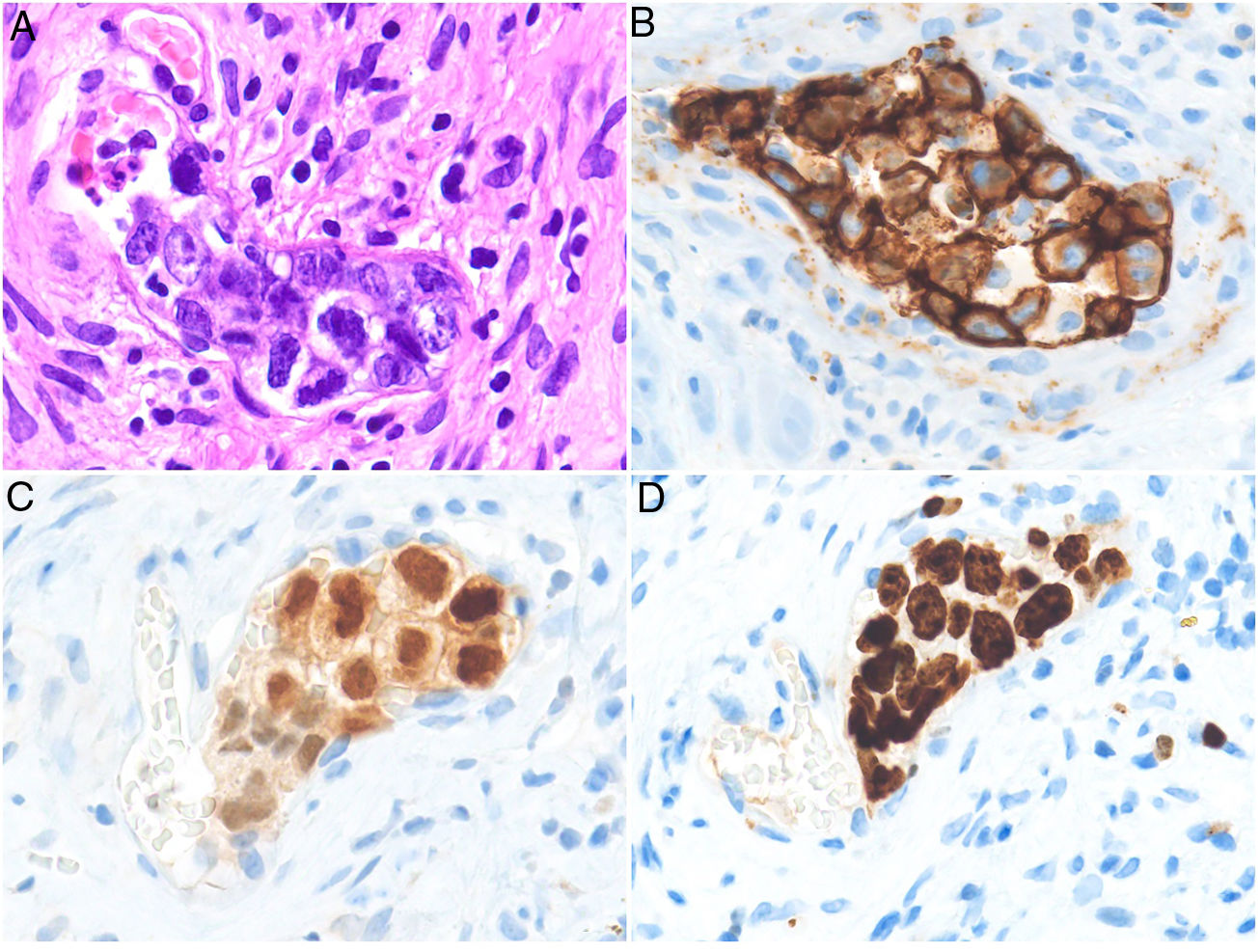
Intravascular large B-cell lymphoma. A, Capillary partially occluded by large blastic-appearing cells. Hematoxylin-eosin, magnification × 400. B–C, Immunohistochemical positivity for CD20 and MUM-1, respectively. Magnification × 400. D, Elevated Ki-67 antigen proliferation. Magnification × 400.
Angiosarcomas are uncommon malignant tumors that arise from the vascular endothelium; they are locally aggressive and able to metastasize.49 Given their capacity to grow inside the blood vessel, they can cause occlusion or emboli.16
Clinical presentationAngiosarcomas are classified as primary or secondary; the latter are related to radiotherapy or chronic lymphedema.49 Clinical signs are edema (of the face in Wilson Jones angiosarcoma) with bruised or congested areas.50 When these tumors cause vascular occlusion, there may be livedoid retiform lesions that mimic vasculitis (Fig. 9A), purpura, or even ulcers and infarcts.16
Histopathology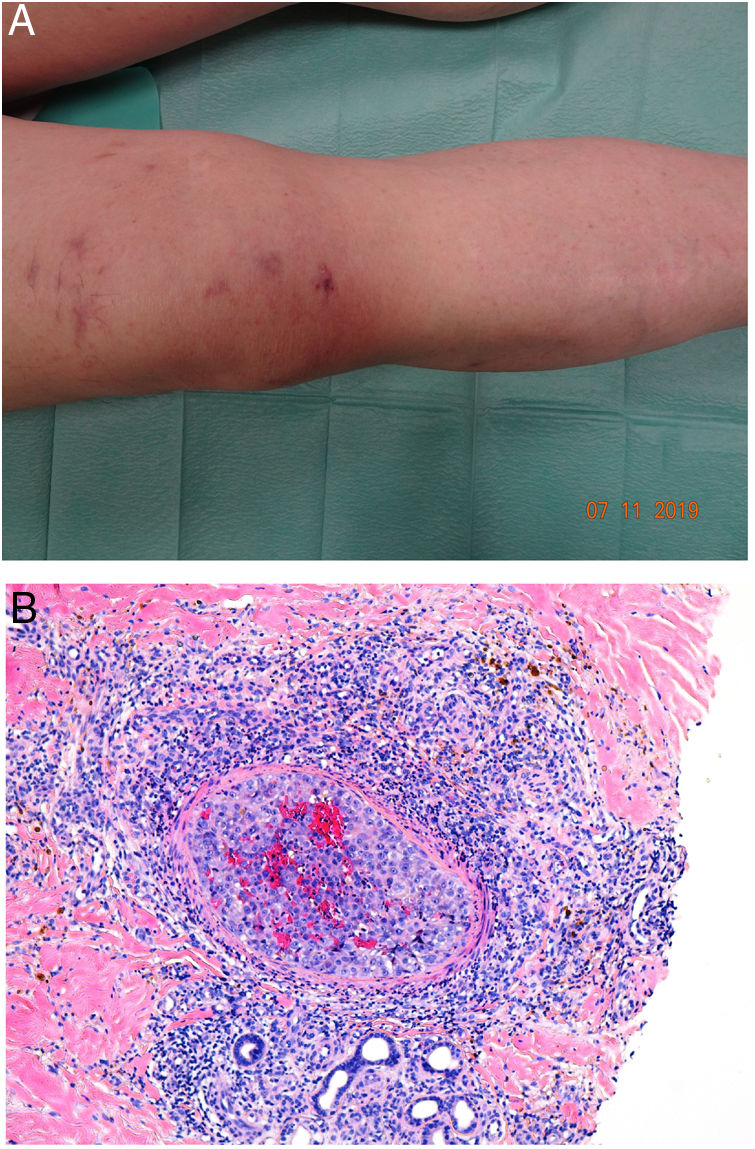
The histologic patterns are highly variable. In well-differentiated angiosarcomas the findings are invasive vascular proliferation with slightly atypical endothelia; in less well-differentiated tumors, the pattern is more solid, and spindle or epithelioid cells predominate.50 Findings of vacuoles in the cytoplasm (corresponding to a rudimentary vascular lumen), red cells, and patches of inflammatory infiltrate will be useful when evaluating solid-pattern angiosarcomas.51
Some variants can display intravascular growth resembling carcinomatous lymphangitis (Fig. 9B). This clinical picture occurs in epithelioid-pattern tumors and in embolization of large vessels due to angiosarcoma.52 It will be useful to demonstrate the immunophenotype of tumor cells with vascular markers (for CD31, CD34, or ERG). Often lymphatic differentiation (podoplanin, D2-40, LYVE-1, or PROX-1) is included.50,51 Bear in mind that in some undifferentiated tumors, the expression of vascular markers is low, but endothelial markers such as MNF116 and cytokeratins AE1/AE3 may be expressed.16,51 In such cases, the intraluminal epithelioid morphology distinguishes an angiosarcoma from an epithelioid hemangioendothelioma, which displays the characteristic WWTR1–CAMTA1 or YAP1-TFE3 translocations.53
Intravascular metastasisDefinitionThe term intravascular cutaneous metastasis refers to the dissemination of a primary tumor by means of lymph or blood vessels.54 Lymphatic spread is much more frequent and has been described as carcinomatous lymphangitis, inflammatory carcinoma, or carcinoma erysipeloides.55 In this paper we are focusing on intravascular occlusion by red cells, so we will use the term telangiectatic carcinoma.56 Hematogenic metastases have been shown to originate with the adherence of tumor cells to the vascular endothelium, followed by cell proliferation and migration; a breach in the vessel wall and invasion of interstitial tissue are unnecessary.57
Clinical presentationAn erythematous plaque with evident telangiectases on the surface can be observed (Fig. 10). The lesion may also take on a pseudovesicular aspect that mimics a circumscribed lymphangioma. Intravascular metastatic lesions are similar to angiosarcomas, especially when they are on the cephalic portion of an extremity or in the region of the breasts.58
Histopathology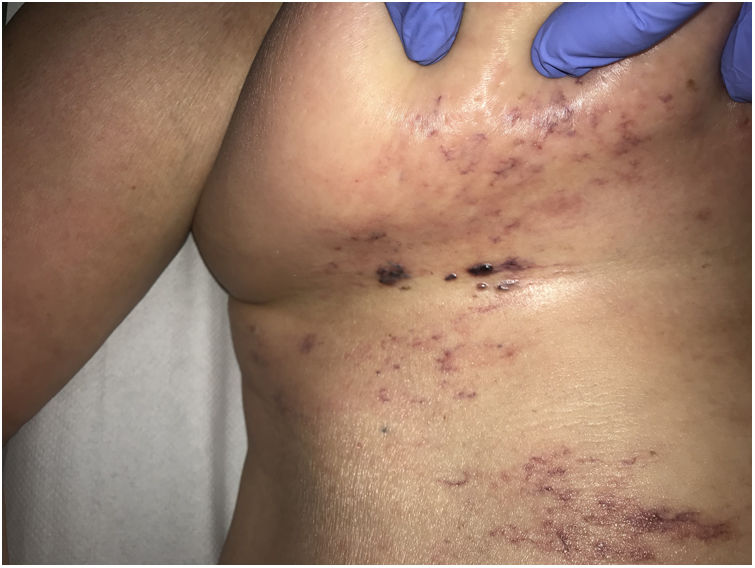
Aggregations of neoplastic cells and red cells can be seen occupying dilated dermal blood vessels. Malignant cells are not found among collagen bundles, nor do they accumulate outside blood vessels. Double staining with CD31 and D2-40 markers can demonstrate that tumor aggregation is confined to blood vessels (CD31+ and D2-40−) (Fig. 11).
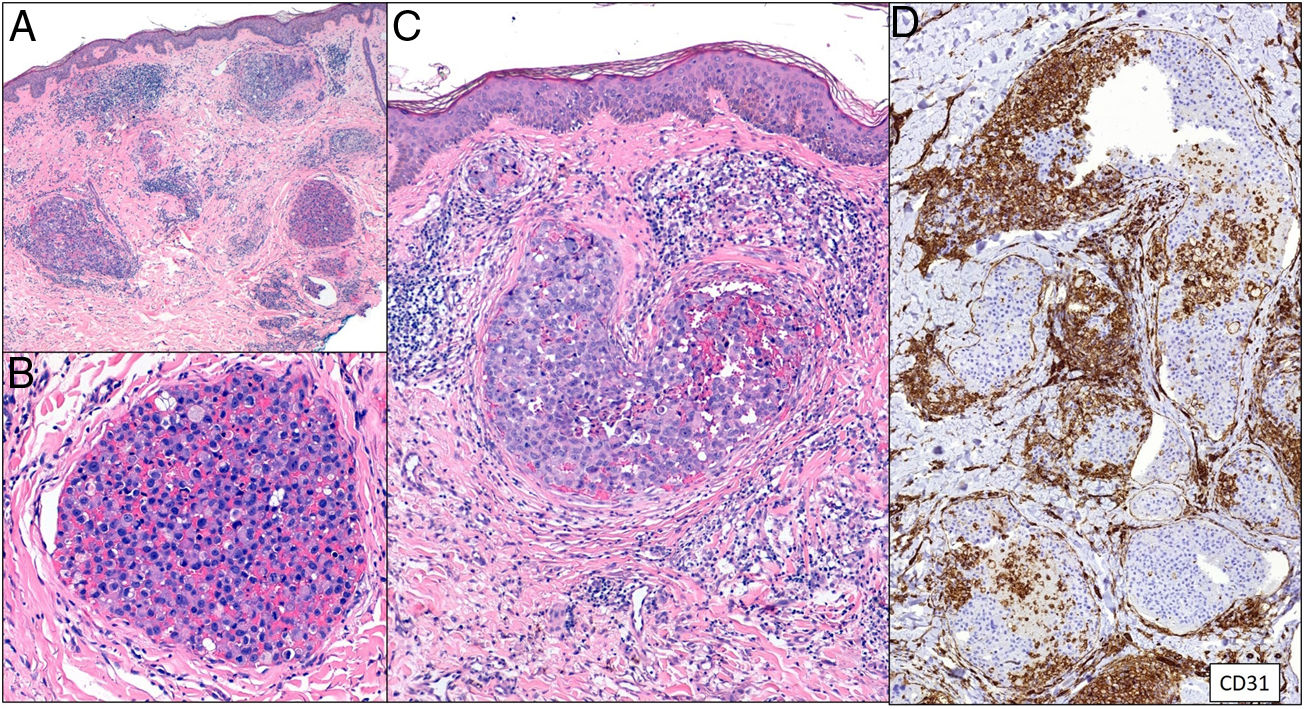
Tumoral thrombi, with dilated vessels throughout the dermis and the vascular lumen completely occupied by pleomorphic cells (mixed epithelioid and red cells). CD31 immunostaining demonstrates the nature of the tumoral proliferation inside the vessel. A, Hematoxylin-eosin (H&E), magnification × 20. B, H&E, magnification × 400. C, H&E, magnification × 200. D, CD31, magnification × 100.
Cutaneous emboli do not form only out of endogenous material. Occasionally the culprit may be foreign material that enters during endovascular procedures or chemotherapy. The hydrophilic or hydrophobic coatings placed on endovascular devices to reduce friction, minimize endothelial damage, and lower the incidence of arterial spasm have been reported to be sources of emboli-forming exogenous material.59 Although foreign-body emboli are relatively rare in the skin, they have been observed more often of late because of the growing number of intravascular diagnostic and therapeutic procedures.
Clinical presentationThe most common sign of foreign body embolism is livedo reticularis, whose multiple causes must be ruled out. Retiform purpuric lesions (Fig. 12), ulcers, and gangrene have also been reported.59–61
Histopathology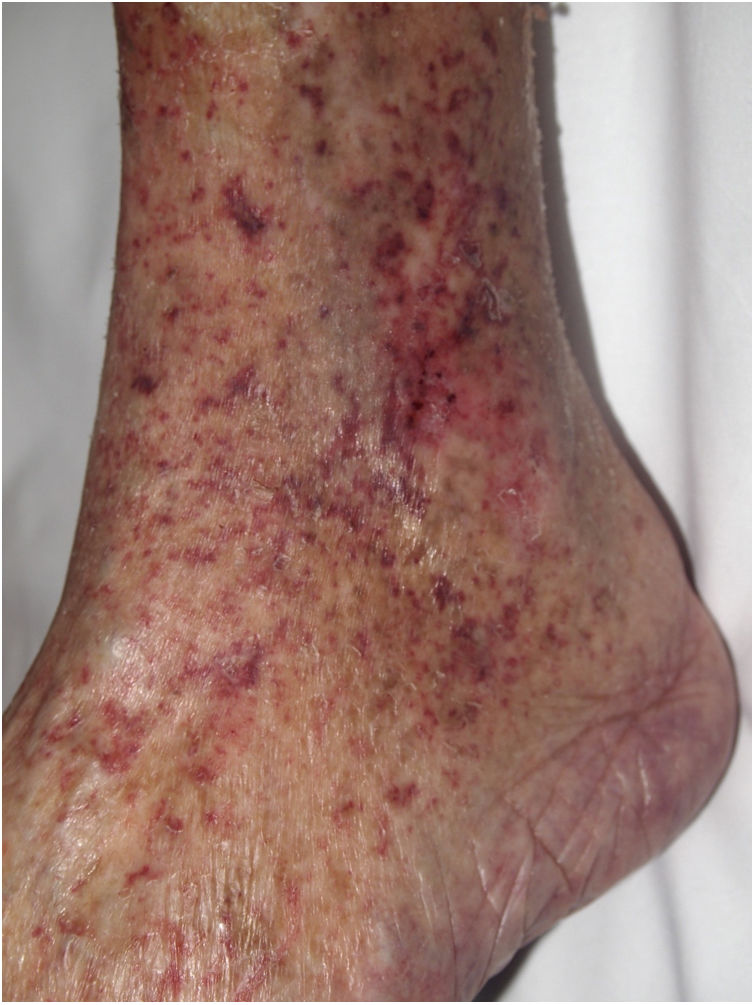
Amorphous or granular basophilic material can be observed in dermal vessels or subcutaneous cellular tissue, often distributed in a characteristic laminar pattern (Fig. 13). Foreign material stains with colloid iron and Alcian blue and is not bright in polarized light. Signs of vascular wall inflammation are characteristically absent, although a certain degree of nonspecific inflammation can be seen in the surrounding dermis.60–62 In differential diagnosis, the most important condition to rule out is cutaneous embolism related to an atrial myxoma, which is characterized by intravascular accumulation of myxoid material in dermal arterioles. However, stellate fibroblasts within the stroma of the embolism, mimicking the structure of the original tumor, facilitate differential diagnosis in most cases; if necessary, immunohistochemical staining for endothelial cell markers will clear up doubts.16 It is very difficult to distinguish other much rarer causes of embolization, such as hyaluronic acid after cosmetic procedures63,64 or intraarticular injections for osteoarthritis.65,66 The patient’s clinical history is therefore of great value.
Emboli caused by microspheres released from doxorubicin agents have also been reported.67 The biopsied tissue in such cases contains purplish, spherical exogenous material inside vessels. In some cases there is also focal extrusion into the adjacent dermis. The conditions to rule out are monoclonal cryoglobulinemia and deep mycosis, but microspheres are rarely difficult to miss because of their perfect shape. A simple technique like PAS staining can occasionally be useful because the microspheres will not stain; in contrast, the thrombi of cryoglobulinemia or fungal infections will.16,67 We can expect the incidence of foreign body emboli to rise given the increase in the number of endovascular treatments.
Finally, other isolated cutaneous embolic complications have been reported in intravenous drug addicts who inject crushed oral medications68 or in patients undergoing arterial embolization with microbeads.69
MiscellaneousWe include a group of miscellaneous causes of vascular occlusion that do not fall into any of the previous groups, whether because they are multifactorial or arise from mechanisms that are different from those explained above.
CalciphylaxisDefinitionCalciphylaxis (or calcifying uremic arthropathy) is a rare, serious complication of unknown etiopathogenesis. It is characterized by the calcification of the media of dermal arterioles and capillaries and subcutaneous cellular tissue. The condition is a characteristic finding in patients with hyperparathyroidism and chronic renal insufficiency undergoing dialysis, although it has also been reported in patients with normal kidney function.70 The patients most often affected are White, female, middle aged or older, diabetic, or infected with the human immunodeficiency virus. Calciphylaxis is associated with high rates of mortality and morbidity.
Clinical presentationThe first lesions are erythematous, livedo reticularis-like signs on the legs. They progress to highly painful purplish lesions in plaques or nodules, affect subcutaneous cellular tissue, and become chronic. Ulcers and necrosis may even develop (Fig. 14A). Several proximal and distal patterns have been described; proximal lesions have a worse prognosis.71
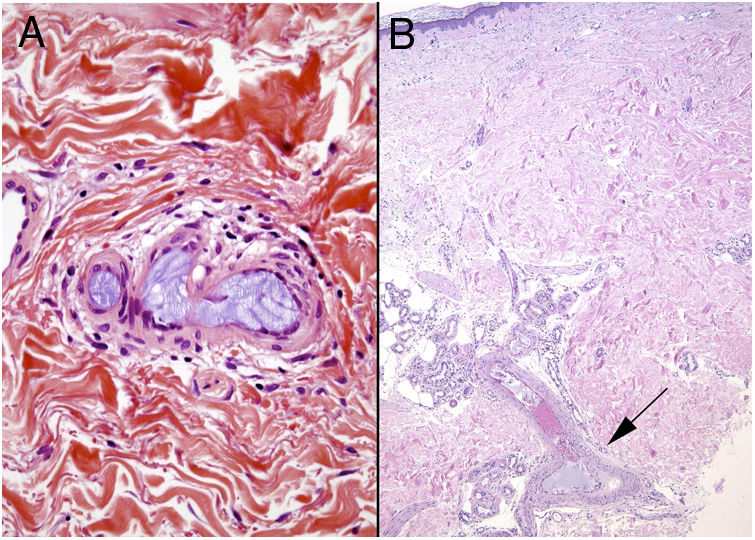
Histopathology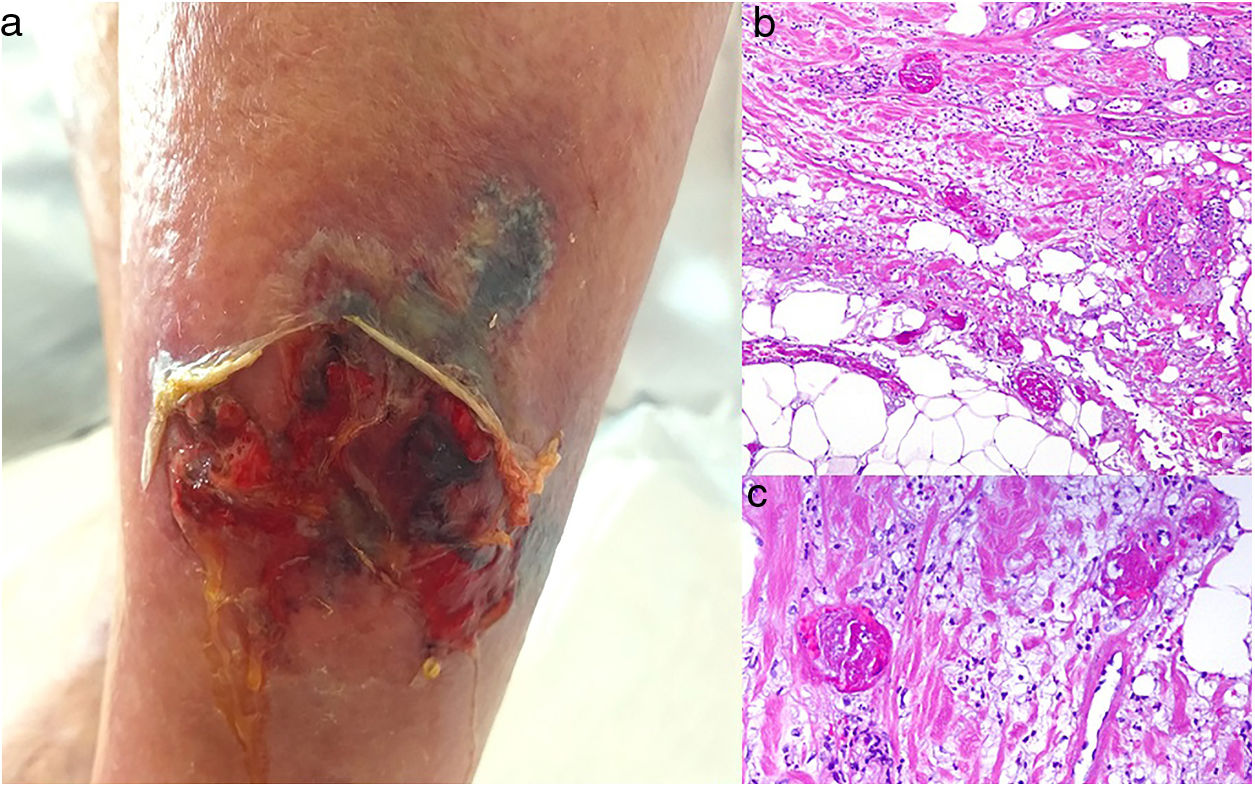
The most characteristic finding is calcification in the media of arteries and arterioles (Figs. 15A–B). Calcifications are accompanied by necrosis and intravascular thrombi72 (Figs. 14B–C). Proliferation in the intima and narrowing of the lumen may also be observed (Figs. 15C–D). Interstitial calcification is an occasional finding (Fig. 15D). Perieccrine calcification has been reported. Rarely, elastic pseudoxanthoma-type changes have been reported.73 It is also common to find areas with extravasated red cells and fat necrosis in subcutaneous cellular tissue, accompanied by a lymphocytic inflammatory infiltrate affecting the lobule.
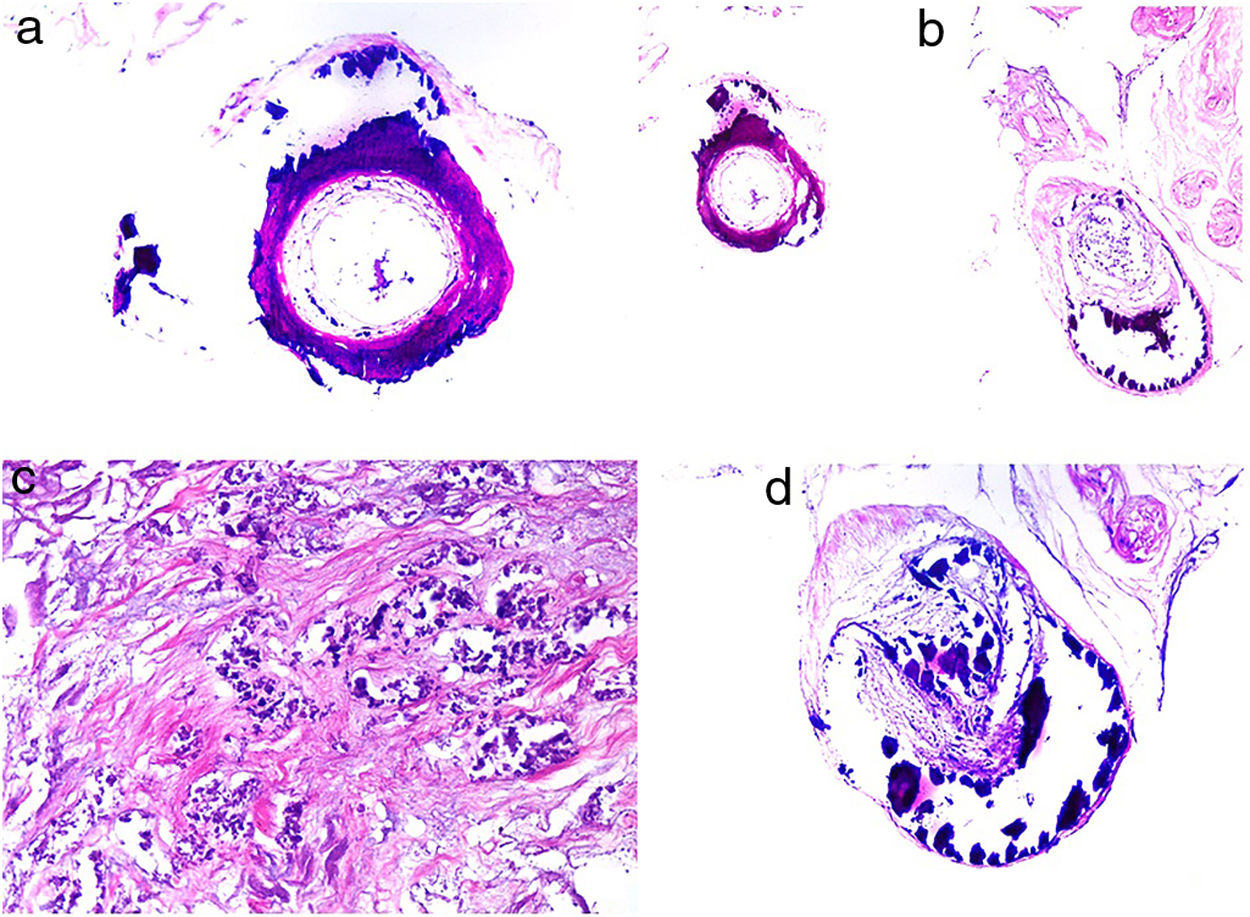
Calciphylaxis. A–B, Calcification of the media of medium-caliber vessels. Hematoxylin-eosin (H&E), magnifications × 100 and × 200, respectively. C, Calcification and interstitial necrosis. H&E, magnification × 400. D, Intimal proliferation accompanied by calcification of the vessel wall. H&E, magnification × 200.
A cumulative dose of at least 510 g after a year’s treatment with the nonalkylating agent hydroxyurea, which blocks the cell cycle in phase S, can cause rheologic changes in red cells that may trigger the coagulation cascade.74,75
Clinical presentationPainful ulcers near the ankle are common. The ulcers are similar to those observed in livedoid vasculopathy and may leave stellate scars.75
HistopathologyFindings are nonspecific. Dermal fibrosis predominates because this is a chronic process, and some blood vessels contain fibrin thrombi. It is almost more typical to find interface dermatitis associated with epidermal atrophy, with occasional apoptotic keratinocytes and hemosiderophages.75
Thrombotic vasculopathy induced by levamisole-laced cocaineDefinitionSince 2005, cocaine has often been laced with levamisole (70% to 90% of the time in the United States). Levamisole is an inexpensive white powder that augments volume and contributes to the stimulant effect of cocaine by raising the level of dopamine in the brain.76 The resulting mixture causes thrombotic adverse events.
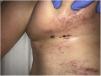
Clinical presentationSkin lesions are the most common clinical component of the vasculopathy syndrome associated with the combination of levamisole and cocaine. The lesions are painful and range from retiform purpura to more confluent purpuric plaques in more than 80% of patients (Figs. 16A and C). Hemorrhagic blisters and necrosis may develop. Lesions affect mainly the upper and lower extremities, the face, and the abdomen, although they may also show a predilection for the ears (> 70% of cases).77
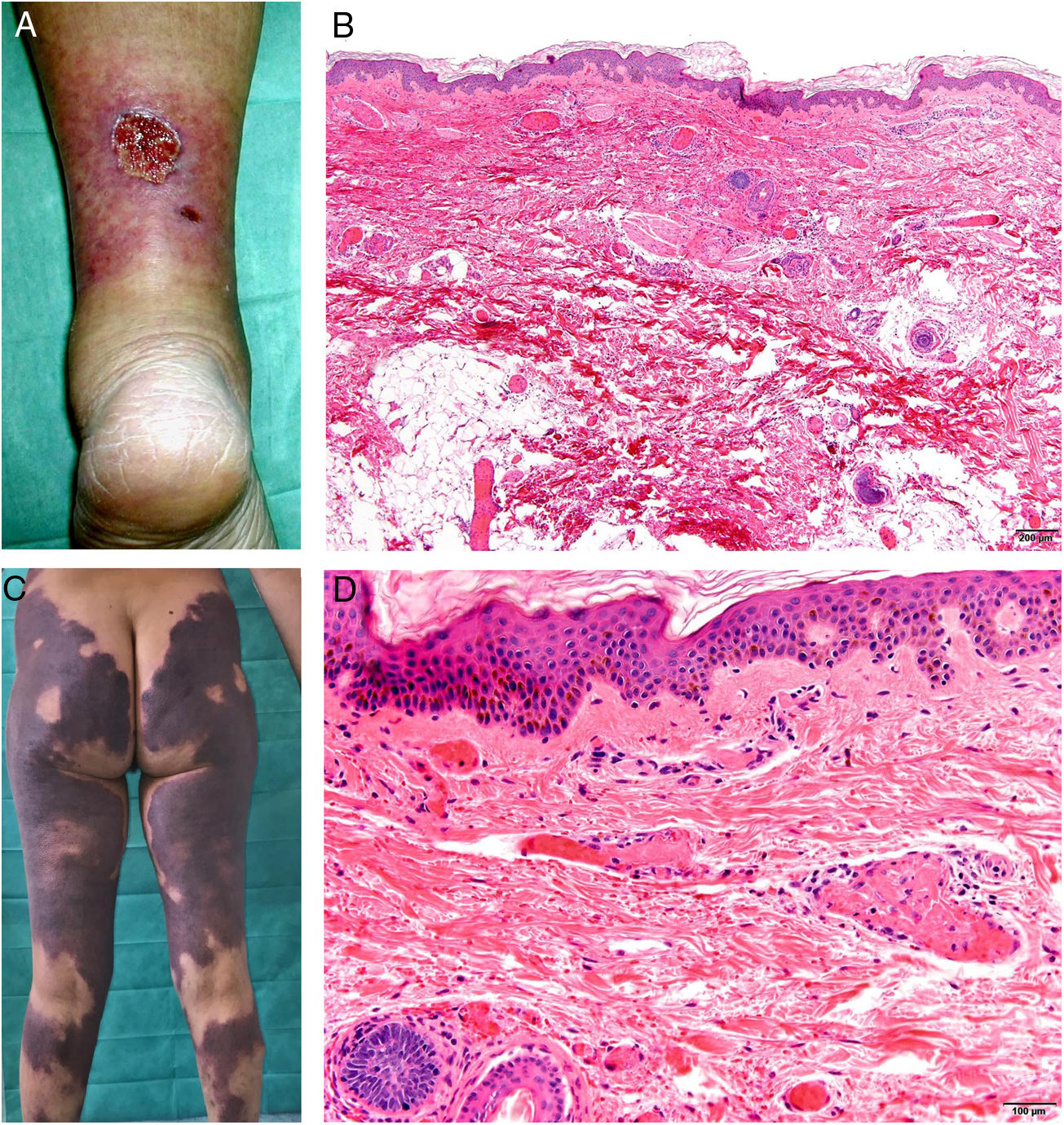
A, Ulcer secondary to hydroxyurea therapy. Photograph courtesy of Dr Fernando Cabo, Dermatology Department, Hospital Universitario de Ourense, Spain. B, In vasculopathy syndrome associated with use of levamisole-laced cocaine, note the intravascular occlusion by fibrin thrombi without vasculitis. Also note the extravasation of red cells and lack of epidermal necrosis. Hematoxylin-eosin (H&E), magnification × 40. C, Confluent purpuric lesions affecting large areas of the lower extremities and buttocks. D, In vasculopathy syndrome associated with use of levamisole-laced cocaine at higher magnification, note the intravascular occlusion with extravasated red cells but without vasculitis. No epidermal necrosis is present. H&E, magnification × 100.
Histologic findings are highly varied, ranging from leukocytoclastic vasculitis and thrombotic vasculitis to occlusive vasculopathy from intravascular fibrin thrombi in the absence of true vasculitis (Figs. 16B and D). Skin ulcers are usually due to severe ischemia.
This review was a project of the Dermatopathology Group of the Spanish Academy of Dermatology and Venereology (AEDV). All the signing authors have contributed equally to the manuscript. The authors are listed in alphabetical order by their first surnames.
Please cite this article as: Beato Merino MJ, Diago A, Fernandez-Flores A. Dermatopatología de la oclusión intraluminal vascular: parte II (coagulopatías, émbolos y miscelánea). ACTAS Dermo-Sifiliográficas 2021;112:103–117.


www.publicationethics.org.
