







INTRODUCCION
Las histiocitosis constituyen un conjunto de procesos derivados de la proliferación de células que, presumiblemente comparten un precursor común CD34+ en médula ósea. La causa de esta multiplicación anómala continúa sin conocerse, barajándose, entre otras, la proliferación clonal, un proceso reactivo mediado por citocinas a estímulos antigénicos desconocidos e incluso participación viral 1 en algunas de las entidades. Presentamos el caso de una paciente que, durante la última fase de tratamiento quimioterápico por una histiocitosis de células de Langerhans (HCL), desarrolló lesiones de xantogranuloma juvenil (XGJ).
CASO CLINICO
Una niña de 2 meses y medio de edad, sin antecedentes personales ni familiares de interés, fue remitida a nuestra consulta por la presencia desde el nacimiento de unas lesiones papulosas eritemato-amarillentas, de superficie costrosa en un número aproximado de 40 a 50, localizadas preferentemente en región cefálica, mitad superior del tronco, axilas e ingles. También se objetivó una pequeña úlcera en la región genital. La niña no tenía otra sintomatología sistémica acompañante. En la exploración se apreciaban múltiples pápulas eritemato-amarillentas, algunas con componente descamativo-costroso, localizadas en cuero cabelludo, mitad superior del tórax y espalda. En ambas ingles tenía placas úlcero-erosivas que se extendían hacia el periné (fig. 1) junto con pequeñas pápulas blanquecinas perianales. No se palpaban adenopatías, masas ni megalias. Una biopsia de una lesión de la espalda mostró un infiltrado epidermotropo que ocupaba y expandía la dermis papilar, que se acompañaba de ulceración y costra central (fig. 2). Dicho infiltrado estaba constituido fundamentalmente por células de núcleo arriñonado con amplios citoplasmas eosinófilos que con técnicas de inmunohistoquímica evidenciaron positividad para S-100 y CD1a (fig. 3). Salpicadas, se observaban células gigantes multinucleadas, y algún eosinófilo aislado.
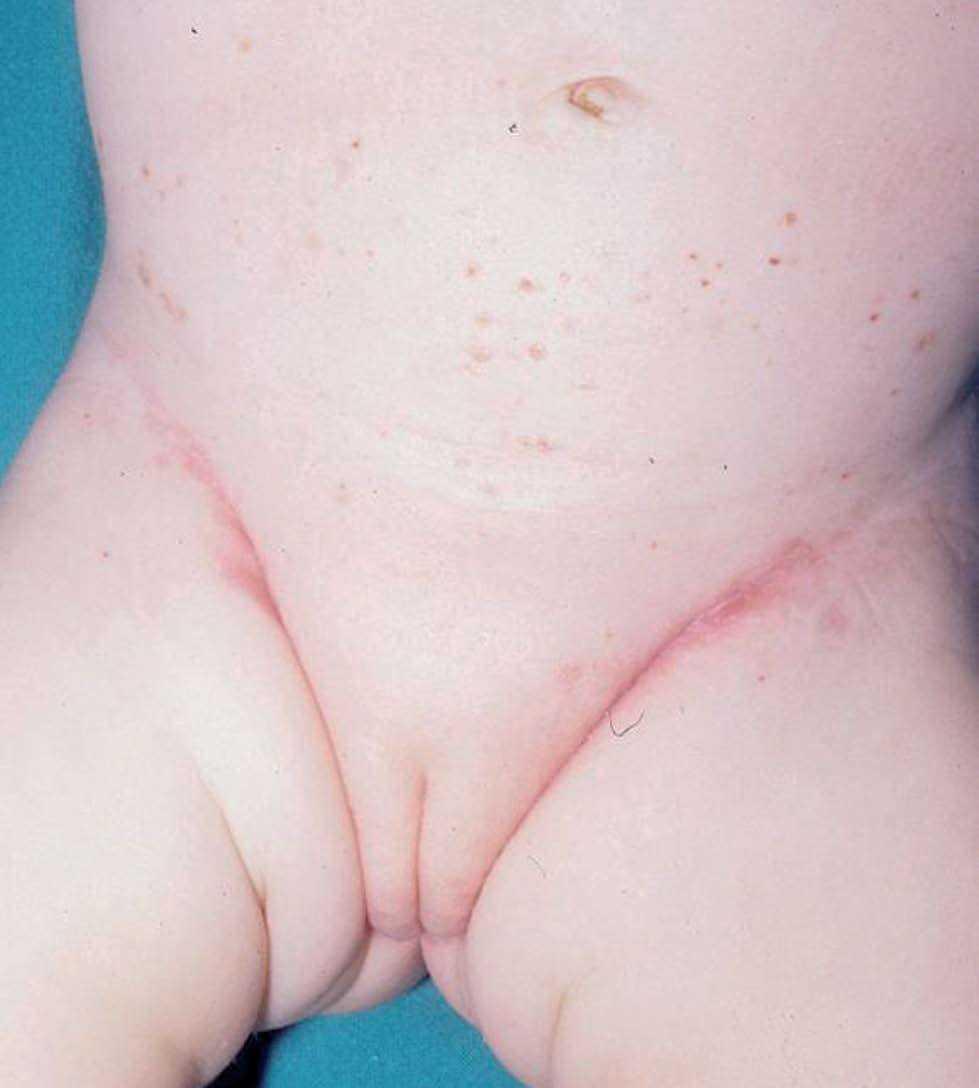
Fig. 1.—Pápulas eritematosas purpúricas y afectación intertriginosa de la histiocitosis de células de Langerhans.
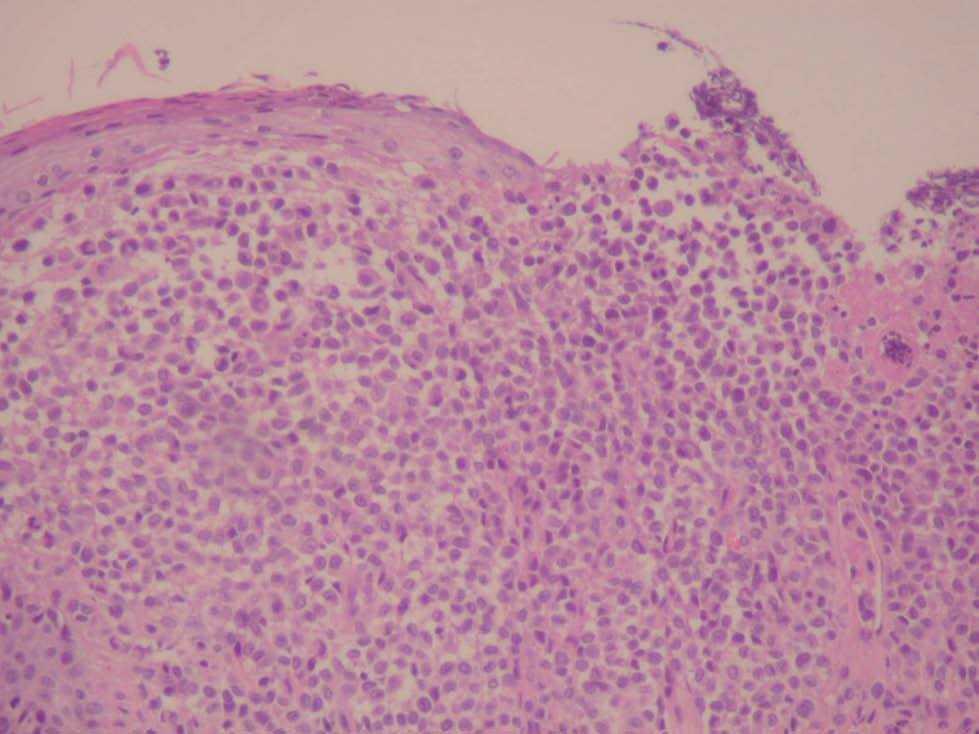
Fig. 2.—Infiltrado dérmico y epidermotropo por células con núcleos arriñonados (hematoxilina-eosina, ×60).
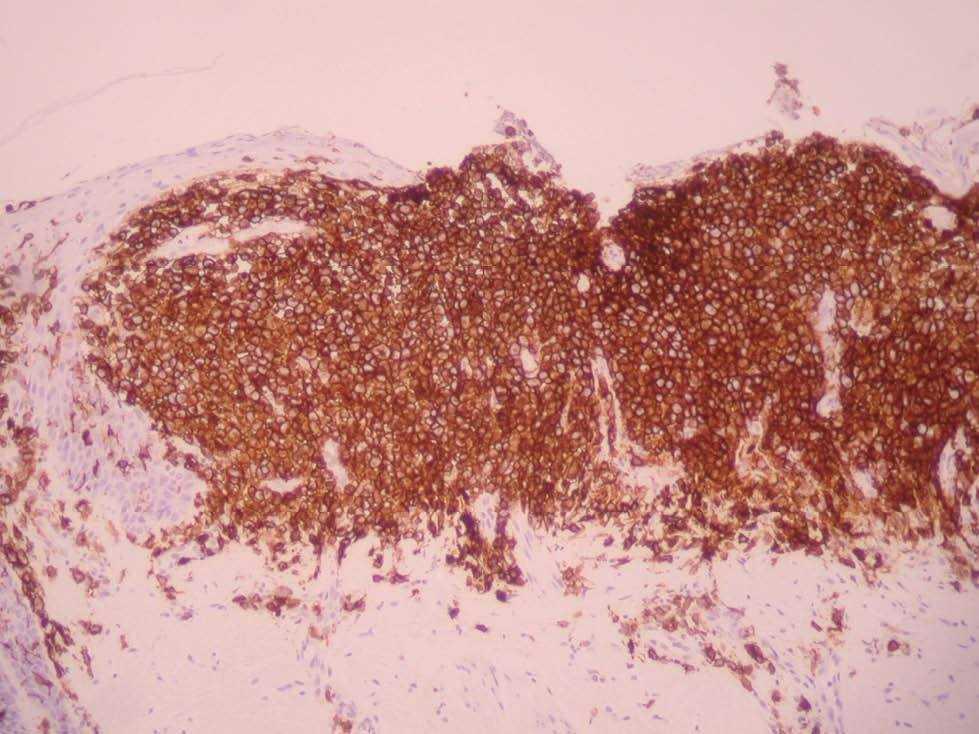
Fig. 3.—Tinción positiva con CD1a.
Se realizaron determinaciones analíticas, en las que destacaba una anemia hipocrómica (hemoglobina 8,9 g/dl), velocidad de sedimentación globular (VSG) 56 mm/h, fibrinógeno 637 mg/dl (normal, 150-400 mg/dl), transferrina 187 mg/dl (normal 230-430 mg/dl) e hierro 15 mg/dl (normal, 33-118 mg/dl). Las inmunoglobulinas, poblaciones linfocitarias y reacción en cadena de la polimerasa (PCR) fueron normales. Las serologías para virus de la hepatitis B (VHB), virus de la hepatitis C (VHC), virus Epstein-Barr (VEB) y virus de la inmunodeficiencia humana (VIH) tuvieron resultados negativos. El estudio de médula ósea mostró buena celularidad global, dentro de parámetros normales. En la ecografía abdominal se visualizó una lesión hipoecoica nodular en lóbulo hepático derecho de unos 11x 7 mm, que posteriormente fue confirmada por resonancia magnética como lesión hipercaptante sugestiva de granuloma dentro de su diagnóstico clínico de histiocitosis. El resto de órganos evaluados (vesícula, bazo, riñones y vejiga) no presentaron alteraciones. En la radiografía lateral de cráneo aparecían dos lesiones de tipo lítico en la región parietal derecha compatibles con HCL. El resto de la serie ósea fue normal. Tras el diagnóstico final de HCL con afectación cutánea, hepática y ósea, la paciente recibió un régimen de tratamiento con prednisona y vinblastina, con mejoría progresiva y desaparición de las lesiones cutáneas. Los estudios de imagen de control a los 6 meses y al año del diagnóstico mostraron remisión completa de las lesiones focales previas en hígado y región parietal derecha.
A las 38 semanas del tratamiento la paciente presentó varias lesiones papulosas semiesféricas eritemato-amarillentas en mejilla derecha (zona preauricular) de pocos milímetros de diámetro, muy bien definidas y tacto sólido (fig. 4). Se tomó una de las lesiones para estudio histopatológico, que mostró un infiltrado difuso en dermis papilar y reticular superficial, constituido en su mayor parte por histiocitos de citoplasma amplio, basófilo, con xantomización ocasional (fig. 5). Se observaban algunos linfocitos y eosinófilos entremezclados. El estudio inmunohistoquímico de estas células mostró un perfil macrofágico CD68+ , S-100 y CD1a (fig. 6). Por todo ello, se llegó al diagnóstico de xantogranuloma juvenil. Durante los meses siguientes al final del tratamiento de la HCL no se observaron lesiones de dicha enfermedad en la piel ni a otros niveles, pero siguieron apareciendo pequeñas pápulas compatibles con XGJ en cara, tronco y miembros.
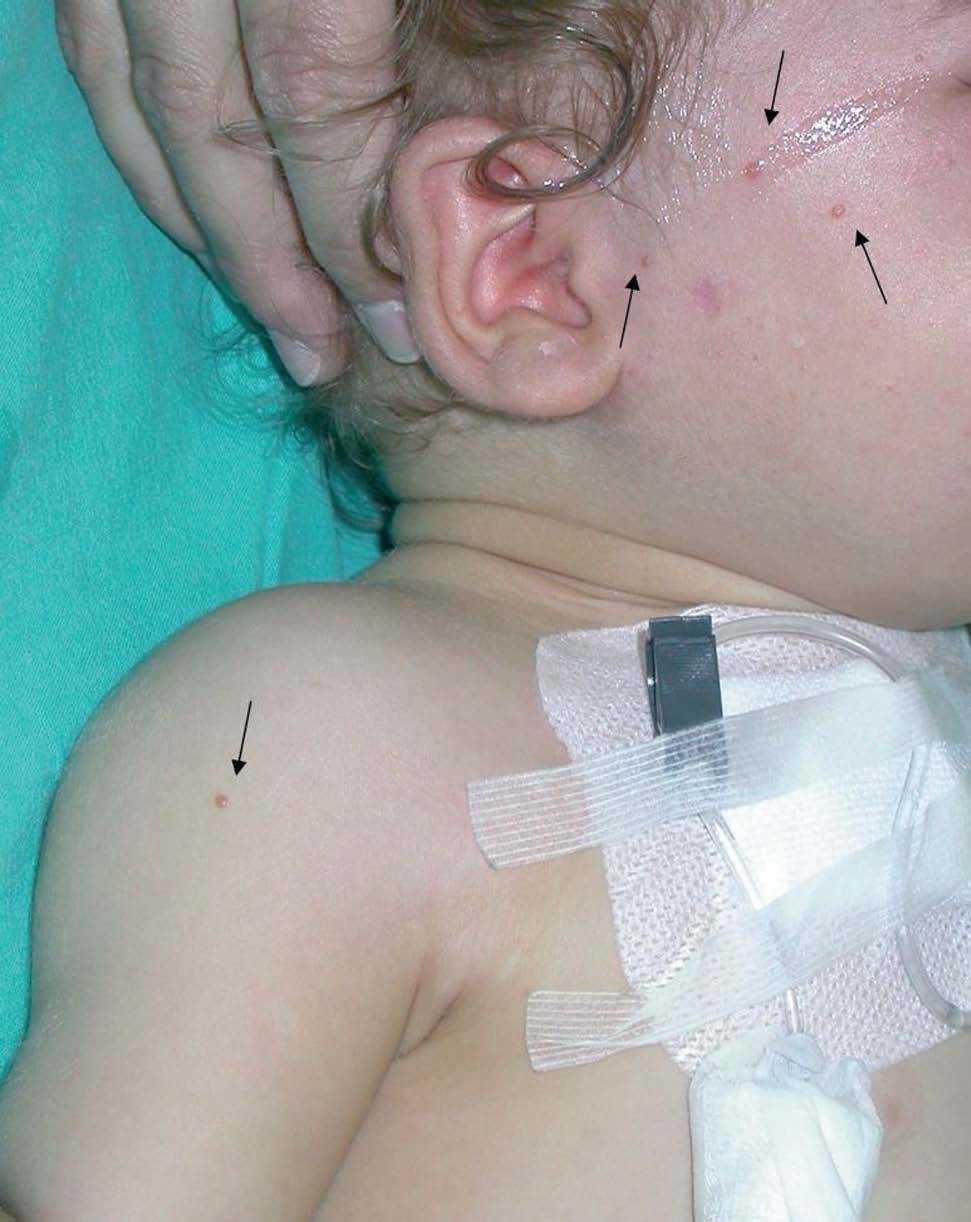
Fig. 4.—Pápulas marronáceas de xantogranuloma juvenil.
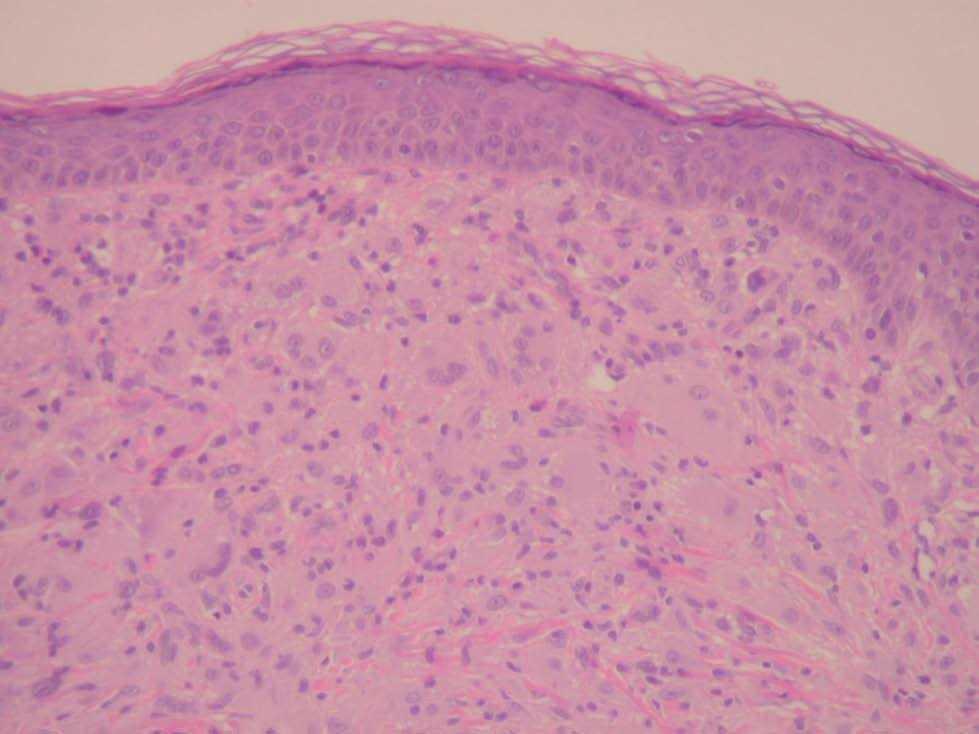
Fig. 5.—Infiltrado dérmico por células histiocitarias xantomizadas formando células gigantes tipo Touton (hematoxilina-eosina, ×60).
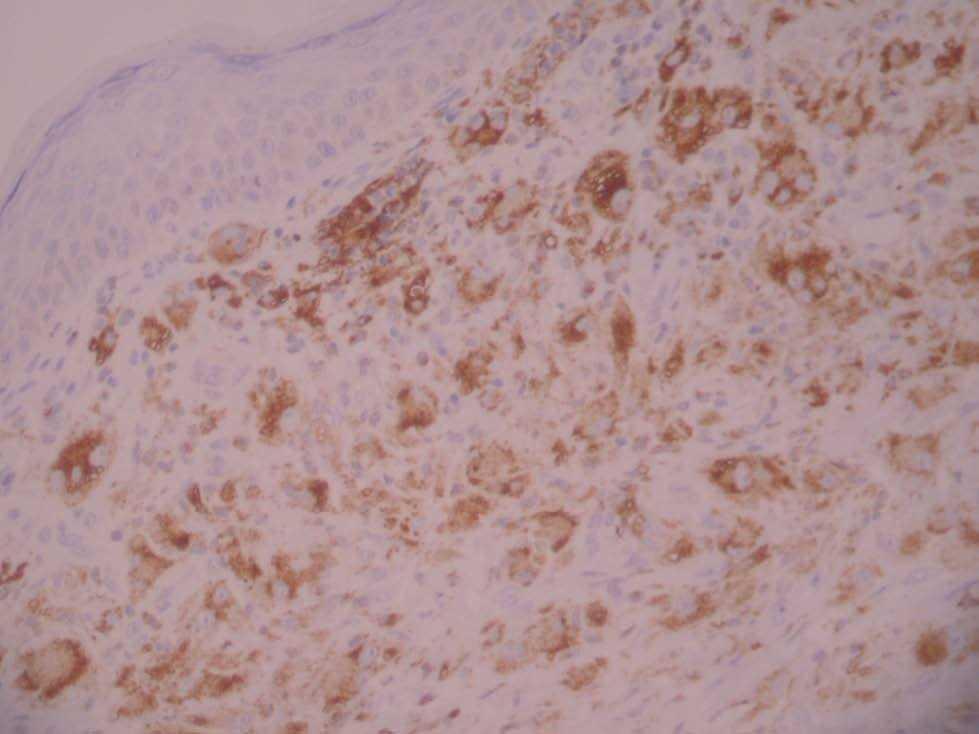
Fig. 6.—Tinción positiva con CD68.
DISCUSION
Existe una íntima relación entre las células causantes de los diferentes tipos de histiocitosis, con superposición en ocasiones de características funcionales, morfológicas y fenotípicas 2. Por éste y otros motivos se ha llegado a pensar que la asociación de varias histiocitosis no es un hecho fortuito; en este sentido algunos autores incluso opinan que dentro de cada «subgrupo» de histiocitosis, entidades clásicamente separadas actualmente deberían ser consideradas como diferentes fases o estadios de un mismo espectro de la enfermedad.
La HCL, comparte con el XGJ algunas características tales como el pico de incidencia en edades tempranas, la localización de las lesiones y la posibilidad de resolución espontánea; aunque la composición celular de las lesiones puede estar influenciada por el tejido en que se localicen, y el estadio de la enfermedad en el momento de la biopsia, habitualmente la histopatología permite una diferenciación clara entre ambos procesos. Los marcadores celulares y la microscopía electrónica son también de ayuda en el diagnóstico diferencial 3.
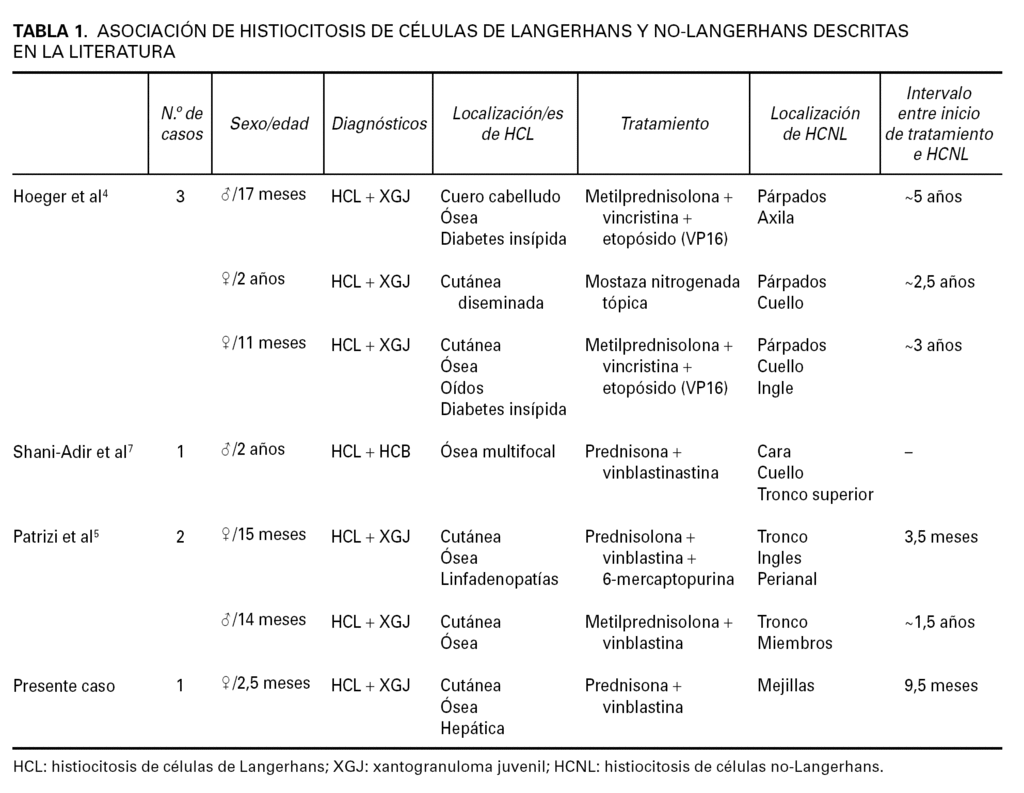
En nuestra paciente, afecta de una HCL desde el nacimiento, aparecieron lesiones de XGJ unos 8 meses después del inicio del tratamiento quimioterápico con vinblastina y prednisona, e incluso continuaron apareciendo XGJ cuando la paciente se encontraba en remisión de la HCL y en ausencia de tratamiento. Hasta la fecha, se han descrito 5 casos previos que asocian HCL con histiocitosis de células no Langerhans (tabla 1). Hoeger et al 4 describieron tres pacientes que desarrollaron XGJ entre 3 y 6 años después del inicio de la HCL. Los XGJ ocurrieron en lugares donde previamente tenían lesiones de HCL, pero no confinadas a ellas. En cuanto a la posible explicación de estos procesos apuntan que los XGJ se han relacionado con una gran variedad de estímulos externos, tales como infección o traumatismo, por lo que la reacción inflamatoria asociada a la HCL podría haber precipitado el desarrollo de XGJ en los pacientes. También se especula acerca del precursor común en médula ósea CD34 + como conexión entre ambas enfermedades. Patrizi et al 5 presentaron dos pacientes, un niño y una niña, de 14 y 15 meses respectivamente, que fueron diagnosticados de HCL, ambos con afectación cutánea y ósea. Fueron tratados sistémicamente con vinblastina, esteroides sistémicos y 6-mercaptopurina y, en un plazo entre 3 y 10 meses, desarrollaron XGJ. Según los autores, una explicación posible para la asociación entre HCL y XGJ es la proliferación clonal y las relaciones entre los histiocitos. En este sentido, dado que el dendrocito dérmico es la principal célula reactiva en el XGJ y que deriva de una célula madre hematopoyética de médula ósea, aquél podría diferenciarse en cualquier tipo de célula dendrítica, incluyendo las células de Langerhans en un ambiente adecuado. La relación y diferenciación entre XGJ e HCL, incluso en el estudio histopatológico, puede ser compleja, porque una lesión temprana de XGJ puede carecer de las células de Touton características y contener escasas células de Langerhans, y por otra parte se ha descrito la xantomización de células en lesiones tardías de HCL como manifestación de la evolución crónica de la misma 6. Sugieren también que la quimioterapia podría modificar la producción de citocinas, estimulando la «maduración» de las células anormales en macrófagos y fusionándolas para formar células gigantes multinucleadas tipo Touton; asimismo, la modificación de las lesiones cutáneas hacia XGJ durante la quimioterapia de la HCL podría ser un factor pronóstico favorable.
Shani-Adir et al 7 publicaron el caso de un niño de 2 años en el que coexistían una histiocitosis cefálica benigna (HCB) y una HCL en forma de múltiples lesiones líticas en cráneo, columna lumbar y tibia, de manera que se apoya la posibilidad de superposición entre distintas histiocitosis. Es bien conocido el amplio solapamiento clínico e histológico entre HCB y XGJ. Weston et al 8 presentaron un caso excepcional de una niña de 5 años con HCB a la que posteriormente se le diagnosticó una diabetes insípida.
Nuestra paciente y los casos referidos resaltan la complejidad de los cuadros de histiocitosis. El origen común de las células y su maduración o activación en un ambiente de citocinas concreto podrían ser la razón principal de este solapamiento, pero no puede descartarse el papel de la inflamación provocada por la HCL o del tratamiento quimioterápico administrado.
Correspondencia:
Antonio Torrelo. Servicio de Dermatología. Hospital del Niño Jesús.
Menéndez Pelayo, 65. 28009 Madrid. España.
atorrelo@aedv.es
Recibido el 27 de febrero de 2006.
Aceptado el 27 de junio de 2006.


