



The patient was a 65-year-old man (a nonsmoker) with a past history of nasal polyps and late-onset bronchial asthma that was very difficult to control. He had been hospitalized multiple times and was a long-term user of oral prednisone at a dosage of 15mg daily. In April 2015, the patient was enrolled by the pulmonology department in a clinical trial that compared lebrikizumab to placebo; the first step was to gradually reduce his prednisone dosage to 2.5mg daily. A week after this dosage was reached, the patient had an asthma exacerbation and very pruritic lesions appeared on the scalp, accompanied 15 days later by paresthesia, pain, and complete hemiplegia of the lower right limb.
Physical ExaminationPhysical examination revealed excoriated, erythematous, purpuric papules and plaques, including some with central ulceration (Fig. 1). No other associated lesions were observed.
Histopathology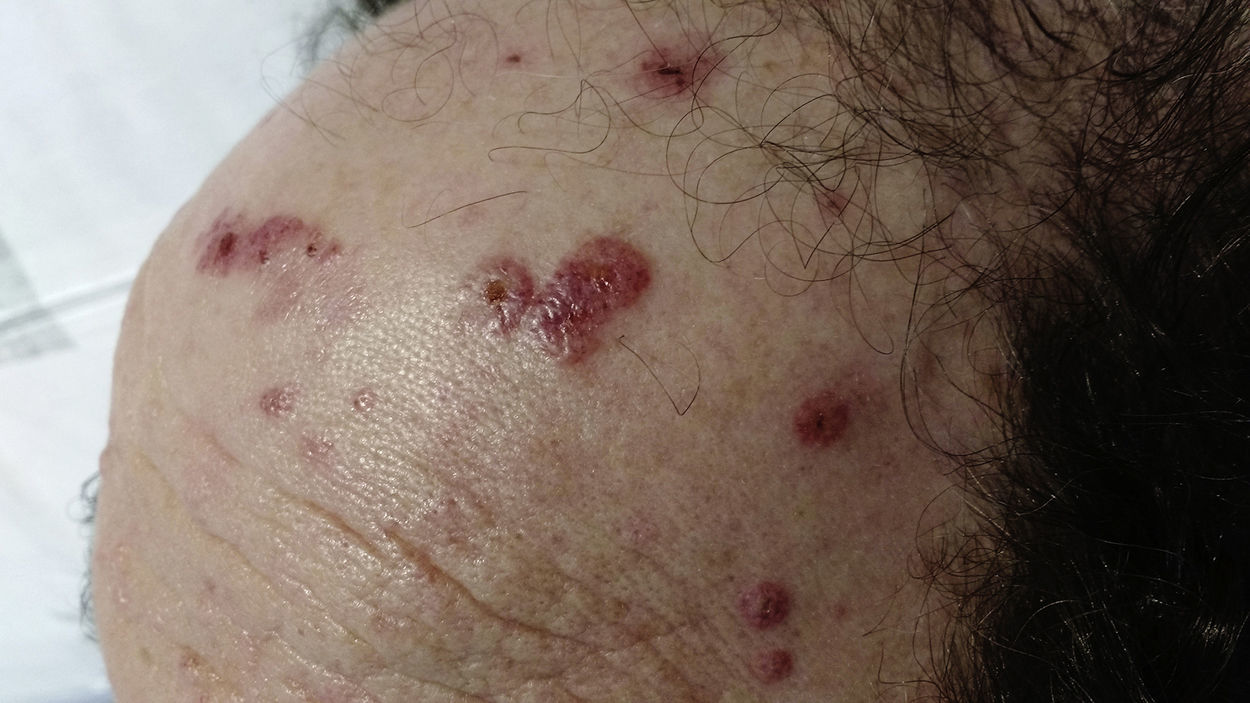
Histologic examination revealed a cutaneous ulcer with necrotizing vasculitis and a mixed neutrophilic and eosinophilic infiltrate in perivascular and interstitial areas (Figs. 2 and 3). Cultures, Gram stain, and direct immunofluorescence were negative.
Additional Tests

Blood tests revealed eosinophilia (19 620 eosinophils/μl; 72%) and perinuclear antineutrophil cytoplasmic antibodies (pANCA) (34IU/mL; <5 IU/mL is normal).
Electromyography revealed mononeuritis multiplex in the right leg.
Chest radiography revealed minimal bilateral pleural effusion.
What Is Your Diagnosis?
DiagnosisEosinophilic granulomatosis with polyangiitis (EGPA).
Clinical Course and TreatmentTreatment was started with intravenous methylprednisolone at a dosage of 1g daily for 5 days, followed by oral prednisone at a dosage of 1mg/kg/d. Adequate response was achieved, except in the neurologic manifestations. The patient was then removed from the clinical trial and treatment was started with rituximab. Significant improvement was observed at subsequent follow-up visits.
CommentEGPA, previously known as Churg-Strauss syndrome, is a multisystem disease that occurs with equal frequency in both sexes and is characterized by allergic rhinitis, asthma, and severe eosinophilia.1 It is classified in the group of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitides affecting small and medium-sized vessels and is the least common of the 3 entities included in this group.1
Given the presence of ANCA, the alteration of type 1 and type 2 T-helper cells, the decrease in regulatory T-cells, and eosinophilia caused by both increased synthesis and decreased apoptosis, it has been suggested that EGPA may have an immunologic pathogenesis.2,3
Several authors have reported EGPA in systemic-corticosteroid-dependent asthmatic patients following a reduction in corticosteroid dosage and the start of treatment with leukotriene modifiers and/or omalizumab.4,5 These authors concluded that the onset of the disease was not related to the start of treatment with new drugs but to the reduction in corticosteroid dosage, and that therefore there was no causal relationship but rather an unmasking of underdiagnosed EGPA.4,5
EGPA develops in 3 phases: a prodromal phase, characterized by underlying atopy and late-onset asthma that is difficult to control; an eosinophilic phase, characterized by prominent blood eosinophilia; and finally a vasculitic phase, characterized by the development of necrotizing vasculitis of small and medium-sized vessels, an eosinophilic infiltrate, and in many cases vascular and extravascular granulomatosis. The vasculitic phase is characterized by constitutional symptoms and multisystemic manifestations such as mononeuritis multiplex, microhematuria, and cardiac failure, which is the most common cause of death.6
Cutaneous manifestations—typical of the vasculitic phase—tend to include subcutaneous nodules, palpable purpura, and hemorrhagic maculopapular lesions. These lesions occur most frequently on the lower limbs; however, lesions on the scalp are distinctive, although not pathognomonic.6
Between 40% and 60% of patients with EGPA are positive for ANCA, typically pANCA, although the presence of these antibodies does not predict activity and should not be taken into account in follow-up.6
There is no specific diagnostic test for EGPA; diagnosis relies on preestablished criteria and evidence of vasculitis in the affected organ.6
Treatment involves systemic corticosteroids, sometimes in association with cyclophosphamide if cardiac, renal, or central nervous system involvement is present.6,7 Rituximab, mepolizumab, and omalizumab have been used in refractory cases, with disparate results.7
Differential diagnosis should include other entities characterized by angiolymphoid hyperplasia with eosinophilia as well as hypereosinophilic syndrome, as these are typically not associated with asthma or histopathologic findings indicative of vasculitis.2,6
In conclusion, we postulate that the reduction in corticosteroid dosage was the factor that triggered systemic manifestations in our patient, since these drugs likely masked the EGPA, which had not yet entered the vasculitic phase. We highlight the characteristic presentation of vasculitic lesions on the scalp. In patients with asthma, severe eosinophilia, systemic manifestations, and skin lesions, early histologic diagnosis is essential in order to begin appropriate treatment.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Villegas FCB, Motilla JMS, Fontestad NR. Vasculitis con lesiones en cuero cabelludo como única manifestación cutánea. Actas Dermosifiliogr. 2018;109:175–176.


