





INTRODUCCION
Las crioglobulinas (CG) son globulinas presentes en el suero que precipitan a temperaturas inferiores a 37° C y se disuelven de nuevo al calentarlo1. Se trata de un fenómeno in vitro, pero el verdadero mecanismo que provoca la crioprecipitación sigue sin conocerse; podría deberse a las características intrínsecas de las inmunoglobulinas o a la interacción entre los distintos componentes del crioprecipitado. Diversas hipótesis han intentado explicar este fenómeno; se ha hablado de modificaciones estructurales en las porciones variables de las cadenas pesadas y ligeras, disminución en la concentración de ácido siálico, disminución de galactosa en la porción de Fc de la Ig4 y mutaciones de las Ig que provocarían la formación de puentes glucosilados en el dominio CH3 2-5. Con frecuencia pueden detectarse pequeñas cantidades de material crioprecipitable en el suero en condiciones normales, sin que tengan ningún significado patológico. Probablemente se deba a interacciones fisiológicas específicas entre las distintas moléculas de las inmunoglobulinas 6.
La primera descripción de crioglobulinemia (CGM) se hizo en el año 1933 por Wintrobe y Buell en una mujer de 56 años que sufría mieloma múltiple y presentaba fenómeno de Raynaud, púrpura, hepatoesplenomegalia y trombosis en venas de la retina 7. Lerner y Watson en 1947 acuñan el término crioglobulinemia 8, y Meltzer y Franklin en 1966 describen la tríada que caracteriza la entidad: púrpura, artralgias y astenia 9.
La etiología en algunos casos de CGM resultaba clara e incluía enfermedades hematológicas, alteraciones autoinmunes y ciertas infecciones (bacterianas, víricas, fúngicas y parasitarias) entre otras 10. Pero existía un gran porcentaje de casos en los que, aunque se sospechaba un posible origen vírico, éste no se conseguía demostrar. No fue hasta la década de los noventa cuando los datos fueron apuntando al virus de la hepatitis C (VHC) como causante de la mayoría de las llamadas crioglobulinemias esenciales. La confirmación de esta relación ha dado un vuelco a la interpretación y el tratamiento de esta entidad.
En este artículo revisaremos el diagnóstico, clasificación y sintomatología de las CGM, y analizaremos el papel del VHC en esta entidad y otras enfermedades relacionadas. Tenemos la gran oportunidad de intentar esclarecer los vínculos que pueden existir entre una infección vírica, diversas alteraciones autoinmunes y alteraciones linfoproliferativas benignas y malignas.
CLASIFICACION
Las CGM pueden ser primarias (esenciales) o secundarias según se asocien o no a algún otro trastorno identificable. Desde el punto de vista de la naturaleza bioquímica de la crioproteína, la clasificación más empleada en la actualidad sigue siendo la propuesta por Brouet et al en 197411, que debido a su simplicidad y a la buena correlación con las distintas manifestaciones clínicas es de gran utilidad práctica. Contempla tres tipos de CGM. La CGM tipo I representa alrededor del 10%-15% del total, la constituyen CG únicas, compuestas íntegramente por inmunoglobulinas (Ig) monoclonales. Se trata normalmente de IgM y con menor frecuencia de fracciones de IgG. Son habitualmente secundarias a gammapatías monoclonales (mieloma múltiple, macroglobulinemia de Waldenström, enfermedades de cadenas pesadas o linfomas). A veces el crioprecipitado está formado sólo por cadenas ligeras y se excretan en la orina como crioproteinuria de Bence Jones. En la CGM tipo II, que constituye alrededor del 60% de los casos, aparecen CG mixtas, con actividad de factor reumatoide (FR) monoclonal. En la mayoría de los casos las Ig involucradas son IgG e IgM, siendo esta última la que con mayor frecuencia posee actividad de FR monoclonal. Estas paraproteinemias son secundarias habitualmente a enfermedades linfo o mieloproliferativas, enfermedades autoinmunes y procesos infecciosos, pero también pueden ser primarias. La CGM tipo III representa el 25% restante, y en ella las CG son policlonales, compuestas por varios tipos de Ig, en los que se puede detectar actividad FR. Pueden ser secundarias a enfermedades autoinmunes, procesos infecciosos o inflamatorios de diversos tipos y también pueden ser primarias. En ocasiones se encuentran CG que no encajan correctamente en ninguno de los grupos previamente descritos; de hecho se ha propuesto añadir a la clasificación de Brouet un subtipo II-III que presentaría un componente IgG policlonal y una fracción IgM oligoclonal. Esta microheterogeneidad en el componente IgM podría representar la transición de un tipo III con una fracción IgM-FR policlonal a un tipo II-III con una fracción IgM-FR oligoclonal, y por último a un tipo II con una fracción IgM-FR monoclonal, demostrándonos que se trata de un fenómeno dinámico y no de compartimentos estanco12.
Se ha demostrado una mayor frecuencia del subtipo IgG3, que parece tener un mayor poder criogénico, tal vez debido a la facilidad para interaccionar entre fracciones Fc-Fc13.
DIAGNOSTICO
El diagnóstico de CGM se hace en el laboratorio y se basa en la presencia de las CG en el suero. La prueba descrita hace más de 50 años por Wintrobe y Buell7 es la piedra angular para establecer el diagnóstico de la entidad. Ciertos anticoagulantes como la heparina y el fibrinógeno pueden causar crioprecipitados que condicionan falsos positivos, por lo que la extracción de sangre se realizará en un tubo sin anticoagulante, previamente calentado a 37° C. De ahí se transfiere a un recipiente a la misma temperatura para que el proceso de coagulación ocurra a la temperatura corporal. Deben transcurrir por lo menos dos horas antes de hacer la separación del suero. Existe una variante de esta prueba, el método de McLeod y Sassetlo diseñado en 198413, que permite investigar simultáneamente CG e hipocrioglobulinas (hipo-CG). Se trata de CG que sólo pueden identificarse cuando el suero se hace hipotónico al mezclarlo en proporción 1:1 con agua destilada. Después de preparar dos tubos para el estudio de las CG, uno con suero y otro con suero hipotónico, incubamos a 4° C durante al menos 72 horas y se investiga el precipitado tras centrifugación (1.000 g durante 10 minutos a 4°). Posteriormente el crioprecipitado se separa, se lava y analiza para determinar el tipo y la cantidad de CG e hipo-CG. Para determinar el tipo se emplean métodos de inmunodifusión o inmunoelectroforesis con sueros anti-IgG, IgM o IgA. Para la cuantificación pueden emplearse métodos colorimétricos o cuantificación del volumen del crioprecipitado (criocrito). Una vez que se ha registrado la presencia de un crioprecipitado es importante recalentarlo a 37° C, ya que muchos sueros normales forman precipitados irreversibles, mientras que las verdaderas CG son nuevamente solubles. El método descrito por McLeod y Sassetlo es dos veces más sensible que los métodos convencionales. El estudio de las hipo-CG es particularmente importante en casos en los que, a pesar de cuadros clínicos sugestivos de CGM, no es posible identificar la crioproteína por el método habitual.
OTROS ESTUDIOS DE LABORATORIO
Los pacientes con CGM tipo I presentan alteraciones de laboratorio secundarias a la enfermedad hematológica de base como anemia y otras citopenias, hipercalcemia, hiperproteinemia, paraproteinemia monoclonal, retención nitrogenada, hiperviscosidad contra otros. Los pacientes con CGM mixta pueden presentar anemia en dos de cada tres casos; la leucopenia y trombocitopenia son habitualmente secundarias al tratamiento. Puede haber anticuerpos antieritrocitarios y antiplaquetarios, y es frecuente la hipergammaglobulinemia. El FR está muy elevado. Las cifras de actividad total del complemento hemolítico se encuentran disminuidas en el 80% de los casos, así como los componentes C3 y C4, siendo a veces el C4 indetectable. La velocidad de sedimentación globular se encuentra anormalmente elevada en el 70% de los casos.
El examen histológico de médula ósea presenta una imagen histopatológica característica con acumulaciones focales de linfocitos y un número aumentado de células plasmáticas14.
MANIFESTACIONES CLINICAS
El cuadro clínico de las CGM es enormemente heterogéneo según el tipo de CGM y la enfermedad de base relacionada con su desarrollo.
La CGM tipo I, como ya hemos comentado previamente, se asocia de manera casi invariable a enfermedades hematológicas linfoproliferativas como mieloma múltiple, macroglobulinemia de Waldenström, enfermedades de cadenas pesadas o linfomas. Dominan los síntomas de la enfermedad de base. Los síntomas de la CGM dependerán del síndrome de hiperviscosidad, propio de cualquier paraproteinemia, que incluye fenómenos vasooclusivos arteriales, he-morragias, insuficiencia cardíaca congestiva, necrosis isquémica distal, úlceras periféricas, fenómeno de Raynaud, cefalea y otros. En general, cuando la CGM tiene un componente monoclonal IgM, las manifestaciones de hiperviscosidad son más severas que cuando se trata de IgG o IgA, con la excepción del subtipo IgG3 y algunas formas de IgA que, dada su tendencia a polimerizar, pueden tener una sintomatología de hiperviscosidad muy florida.
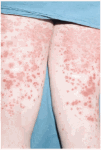
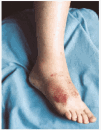
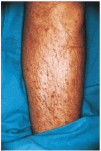
La CGM mixta (tipo II y III) se comporta como una enfermedad mediada por complejos inmunes (CI), capaces de activar el sistema del complemento, tanto por la vía clásica como por la vía alternativa, lo que explica la alta frecuencia de hipocomplementemia en estos pacientes, como ya hemos comentado. Los pacientes podrán presentar la sintomatología típica de una vasculitis sistémica mediada por CI (tabla 1), además de los propios de la enfermedad de base. Como comentamos con anterioridad, son secundarias habitualmente a enfermedades linfo o mieloproliferativas, enfermedades autoinmunes y procesos infecciosos, pero también pueden ser primarias. Con mucha frecuencia las lesiones cutáneas o los fenómenos vasomotores son las manifestaciones iniciales de la CGM. La púrpura es, sin lugar a dudas, la manifestación más frecuente y característica. Se trata de brotes recurrentes de lesiones purpúricas palpables, que comienzan en miembros inferiores, pero que pueden afectar tronco, extremidades superiores y más raramente la cara (figs. 1, 2 y 3). Los brotes pueden precederse de sensación de picor, pero no de auténtico dolor. Duran de 3 a 10 días y dejan huellas de hiperpigmentación. En uno de cada tres pacientes aparecen úlceras en piernas, casi siempre asociadas a púrpura, que frecuentemente se localizan en los maléolos10.

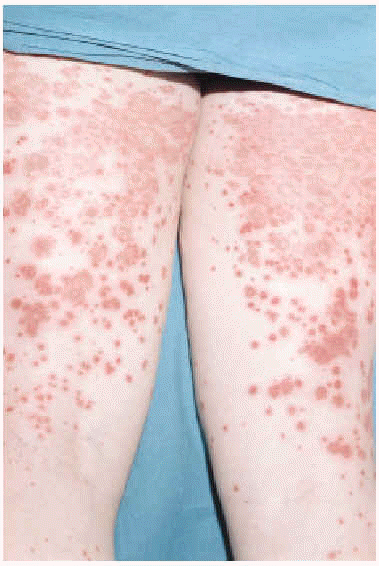
Fig. 1.--Lesiones purpúricas en miembros inferiores de un paciente con crioglobulinemia tipo II e infección por virus de la hepatitis C de reciente aparición.
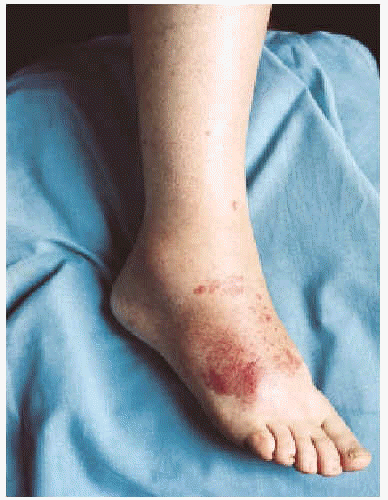
Fig. 2.--Lesiones purpúricas en dorso del pie en paciente con crioglobulinemia tipo III e infección por virus de la hepatitis C.
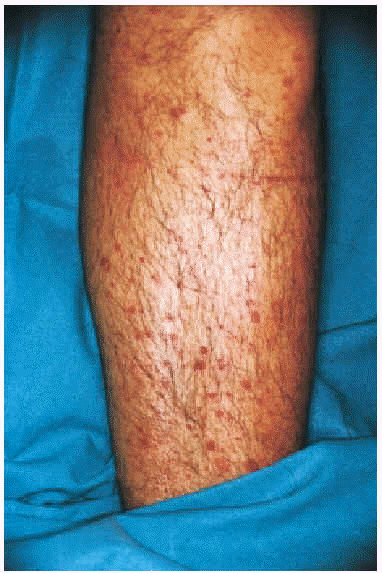
Fig. 3.--Paciente varón con crioglobulinemia tipo II, infección por el virus de la hepatitis C y linfoma de células B no filiado con lesiones purpúricas en tronco y zona axilar.
El fenómeno de Raynaud es también muy frecuente. Afecta generalmente a manos y pies, pero también puede afectar a las orejas, los labios y la nariz. En ocasiones es la manifestación inicial de la CGM.
Los síntomas articulares se presentan en más del 50%. Ocurren preferentemente en manos (interfalángicas y metacarpofalángicas), rodillas y con menos frecuencia en tobillos y codos14. La distribución de las artralgias es simétrica y no migratoria, y no suele asociarse a una verdadera artritis.
La afectación del sistema nervioso varía en la literatura, pero para algunos supera el 50%. Las manifestaciones neurológicas son primordialmente de distribución periférica, y podemos encontrar neuropatías distales de carácter leve probablemente secundarias a infartos de la microcirculación (vasa nervorum), con déficit neurológico muy discreto y mononeuritis múltiples, neuropatías sensitivas y motoras graves asociadas con frecuencia a vasculitis cutáneas necrotizantes que afectan principalmente a los miembros inferiores. La afectación del sistema nervioso central es excepcional14,15.
La afectación hepática la encontramos en casi un 70% de los pacientes diagnosticados de CGM16,17. El estudio anatomopatológico mediante biopsia hepática transcutánea revela una gran variedad de alteraciones, que van desde cambios mínimos de microesteatosis, agregados linfoides portales, hiperplasia de las células sinusoidales, focos de agregados linfomonocitarios intralobulares y necrosis celular intralobular, hasta imágenes de hepatitis crónica activa franca con o sin cirrosis. Dos tercios de los pacientes tienen algún dato analítico que hace sospechar la afectación hepática, como aumento de la fosfatasa alcalina, aumento de las transaminasas, u otros.
La nefropatía varía según las estadísticas entre un 20%-40% de los casos. La manifestación más frecuente es la proteinuria de grado variable, seguida de hematuria, leucocituria, edema e hipertensión. El cuadro histológico más frecuente corresponde a una glomerulonefritis membranoproliferativa18.
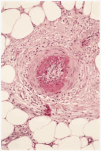
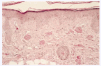
Desde el punto de vista anatomopatológico podemos encontrar hallazgos histológicos típicos de vasculitis leucocitoclástica o signos de poliarteritis nudosa (PAN). El momento más adecuado para realizar la biopsia es a las 24 a 48 horas de la aparición de las lesiones. En la vasculitis leucocitoclástica hay afectación de vaso pequeño (arteriolas, vénulas y capilares) y la arquitectura vascular está alterada por un infiltrado inflamatorio mixto que rodea los vasos e incluso penetra en ellos (fig. 4). Las células endoteliales se muestran prominentes y la presencia de leucocitoclasia y extravasación hemática puede completar el cuadro. Debemos recordar que la leucocitoclasia es un dato no específico, que simplemente revela un infiltrado neutrofílico importante y puede verse en otras entidades como el síndrome de Sweet o diversas infecciones cutáneas. La PAN afecta vasos medianos y pequeños, con presencia de necrosis fibrinoide y un infiltrado inflamatorio mixto de monocitos, linfocitos y polimorfonucleares neutrófilos que interrumpen y borran la arquitectura vascular (fig. 5)19. La presencia de trombos de aspecto hialino, eosinófilos, PAS positivos, ocluyendo la luz de los vasos, aunque muy típica, no es muy frecuente salvo en la CGM tipo I (fig. 6), en la que coinciden con criocritos muy elevados, mientras que las alteraciones de vasculitis, especialmente en las CGM tipo II, son más frecuentes con criocritos bajos. Este fenómeno continúa aún sin explicación, aunque parece deberse a la capacidad intrínseca de los inmunocomplejos de activar el complemento in situ.
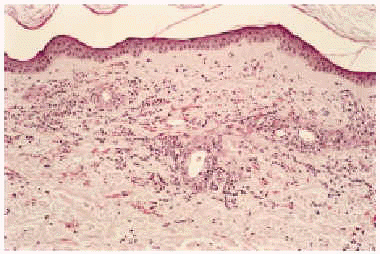
Fig. 4.--Típicos hallazgos histológicos de vasculitis leucocitoclástica.

Fig. 5.--Cambios histológicos diagnósticos de panarteritis nodosa en paciente con crioglobulinemia tipo II e infección por virus de la hepatitis C.
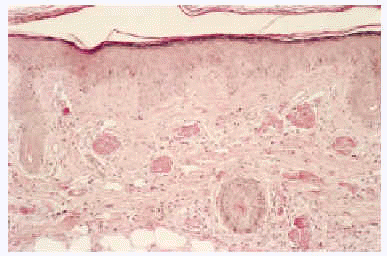
Fig. 6.--Vasos ocluidos por trombos eosinófilos de aspecto hialino, característicos de las crioglobulinemias.
CRIOGLOBULINEMIA Y VIRUS DE LA HEPATITIS C
Dado que en el curso de la CGM es muy frecuente que se desarrolle una hepatitis crónica, desde hace tiempo se venía sugiriendo un posible papel de virus hepatotropos en la patogenia de la enfermedad. En un principio se sospechó del virus de la hepatitis B20, alentados por la asociación de este virus con otro tipo de vasculitis, la PAN que había quedado constatada en la década de los setenta21. Sin embargo, esta sospecha no llegó a confirmarse más que en un pequeño porcentaje de los casos (menos de un 5%). Tras el descubrimiento del VHC como el mayor responsable de las hepatitis no A-no B, las investigaciones se dirigieron hacia este nuevo virus. Numerosos estudios epidemiológicos sugerían un papel importante del virus en la patogenia de la enfermedad22.
El VHC es un virus ARN de cadena simple y una nucleocápside que está dentro de una envoltura formada por las membranas del huésped en la que se insertan diversas glucoproteínas del virus. Su estructura es típica de los flavivirus23. Es el causante de la mayor parte de las hepatitis agudas y crónicas. Se contrae principalmente por vía parenteral (transfusiones, adictos a drogas intravenosas, profesionales médicos, transmisión sexual y, en mucho menor grado, transmisión vertical). Se calcula que el 3% de la población mundial está infectada por el VHC. Además, la incidencia anual estimada de nuevos casos de infección por VHC es de 28.000 nuevos casos por año23.
La sospecha de una posible relación entre la CGM y el VHC se apoyaba en que la prevalencia de anticuerpos anti-VHC en pacientes con CGM era muy elevada, si bien sorprendía que aunque la infección por VHC se distribuía de manera bastante homogénea en el mundo, el desarrollo de CGM asociada al VHC presentaba gran heterogeneidad geográfica24. Esto parece indicar que determinados genotipos puedan ser los responsables, y que factores genéticos y medioambientales deban actuar como cofactores para el desarrollo de la enfermedad25. Actualmente siguen siendo desconocidos esos posibles factores coadyuvantes. Se ha relacionado el HLA-DR11 como posible factor predictivo y coadyuvante del posible desarrollo de vasculitis crioglobulinémica en pacientes infectados por el virus. Asimismo parece que el HLA-DR7 protege frente al desarrollo de la misma26.
Además de los datos epidemiológicos que acabamos de comentar, la relación entre el VHC y la CGM viene avalada por una serie de hechos. La alta prevalencia de ARN-VHC en pacientes con CGM, con una concentración del genoma del virus mucho mayor en el crioprecipitado que en el sobrenadante27. Sin embargo, esta correlación no se demuestra con los títulos de anticuerpos que son iguales en las dos fases. Esto parece indicar que hay un exceso de anticuerpos en relación con las partículas víricas y las proteínas víricas. El crioprecipitado también contiene la mayoría de los antígenos conocidos (core, E1, E2, NS3, NS4, NS5) y sus anticuerpos correspondientes. Por otra parte, la IgG diana del factor reumatoide va dirigida contra proteínas de VHC. Es interesante conocer que la actividad anti-VHC en los complejos inmunes va dirigida tanto contra las proteínas de la superficie del virus como contra las enmascaradas por la cápside28. Finalmente, se ha detectado ARN del VHC en tejidos dañados por la enfermedad como piel y riñón mediante técnicas de inmunohistoquímica e hibridaciónin situ, lo que avala un papel directo del virus en la formación de los complejos inmunes responsables del daño de los distintos órganos en la CGM29, 30.
VHC Y AUTOINMUNIDAD
En los individuos infectados por el VHC se ha observado un gran número de alteraciones inmunitarias que apuntan a que el virus sea el responsable de distintas enfermedades inmunes hepáticas y extrahepáticas. En la tabla 2 se recogen las principales enfermedades que han sido asociadas con este virus. Las enfermedades autoinmunes tiroideas son la enfermedad más frecuente en estos pacientes, especialmente la enfermedad de Hashimoto. La sialoadenitis no se asocia al síndrome de Sjögren típico, pues no presenta xeroftalmia ni anticuerpos anti-Ro (SSA).

También en pacientes afectos de púrpura trombocitopénica autoinmune se ha encontrado una prevalencia alta de VHC.
Los autoanticuerpos son muy frecuentes en estos pacientes. Se detectan anticuerpos antinucleares, contra la porción Fc de la IgG (FR), anticardiolipinas, antimúsculo liso o antitiroideos en el 40% a 65% de los afectados31, 32. Estos anticuerpos se detectan a títulos bajos, salvo el FR y no parecen influir en la presentación clínica de la enfermedad, en el curso de la infección, ni en la respuesta al tratamiento, aunque en ocasiones pueden conducir a errores diagnósticos. Si bien la presencia de anticuerpos autoinmunes no se considera contraindicación para la terapia antivírica, sí se sabe que los títulos de anticuerpos pueden empeorar durante el tratamiento con interferón. Ciertas enfermedades autoinmunes pueden exacerbarse durante el citado tratamiento.
ALTERACIONES LINFOPROLIFERATIVAS Y VHC
Se han publicado numerosos artículos que asocian la infección por VHC con el desarrollo de linfomas no Hodgkin (LNH) de células B33, 34. La prevalencia de este tipo de linfomas es mucho mayor en individuos infectados por el virus que en la población sana; asimismo, la prevalencia de infección por VHC es mucho mayor en pacientes con LNH que en poblaciones con otras enfermedades linfoproliferativas o que la población control. Se han identificado dos tipos de LNH asociado a la infección por el VHC, los LNH en pacientes con CGM, que afectan a la médula ósea y tienen un curso relativamente benigno, aunque ocasionalmente evolucionan a un fenotipo agresivo, y los LNH no asociados a CGM, que no afectan la médula ósea y suelen ser muy agresivos desde el inicio.
Se calcula que los linfomas francos aparecen en un 5%-10% de los pacientes con CGM y VHC35.
El VHC es un virus hepatotropo y linfotropo. Se encuentra de forma activa o latente en los linfocitos periféricos de pacientes con hepatitis C36. Probablemente la infección de tejidos linfoides represente un reservorio para el VHC que contribuye a la persistencia del mismo, pero además las múltiples cuasiespecies del virus le permiten escapar de la vigilancia inmunitaria. Por otro lado se han detectado proteínas y partículas replicativas del virus en linfocitos periféricos y en biopsias de tejidos de pacientes que sufren linfomas mediante inmunohistoquímica, reacción en cadena de la polimerasa (PCR) e hibridaciónin situ37.
A pesar de estos datos, la relación sigue siendo tema de debate. Existe una gran variabilidad geográfica en el desarrollo de LNH en pacientes con VHC, lo que contrasta con la distribución homogénea de la infección por el virus en las distintas áreas. Ello indica que deben existir otros factores, genéticos o ambientales, que influyan en el desarrollo del linfoma. Además, el VHC no necesita para su replicación una cadena intermedia de ADN, por lo que las secuencias víricas del genoma no se pueden integrar en el genoma del huésped. El mecanismo, por tanto, sería a través de un estímulo crónico sobre el sistema inmune, que facilitaría el desarrollo y selección de clones anormales.
Los pacientes con CGM presentan con relativa frecuencia infiltrados linfoides en hígado y en médula ósea, que algunos autores han denominado «linfomas tempranos». Éstos, a diferencia de los linfomas verdaderos, tienden a permanecer estables durante décadas y sólo en un 10% de los casos evolucionan a auténticos tumores. Pensamos que el nombre de trastorno linfoproliferativo de significado incierto es más adecuado para estos casos.
POSIBLE ETIOPATOGENIA DE LA CGM Y OTRAS ENFERMEDADES RELACIONADAS CON EL VHC
La infección del VHC tiene una distribución geográfica muy homogénea; sin embargo, no ocurre así con el desarrollo de la CGM y otras enfermedades relacionadas. Por ello deben existir otros factores implicados en la etiopatogenia que actúen como coadyuvantes y que desencadenen la serie de eventos que a continuación vamos a comentar.
Se acepta de forma general que el virus permanece indefinidamente en el huésped de manera latente o en fase muy lenta de replicación. Se han detectado en el laboratorio secuencias de genoma en ausencia de proteínas víricas en las células B de pacientes con CGM, mientras que no ocurre así en pacientes no crioglobulinémicos. Esto demuestra que estas células no permiten la infección por VHC y su replicación, con lo cual la progresión maligna no está directamente relacionada con el virus38.
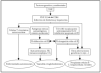
Parece que la proteína E2, situada en la envoltura del virus, interacciona con la proteína CD81, presente tanto en linfocitos como hepatocitos39. La CD81 es una proteína de superficie que está compuesta por CD21, CD19 y Leu 13. Por otro lado, es frecuente encontrar la traslocación T (14;18)40 en los individuos infectados y con CGM. La activación del protooncogén bcl2 llevaría a la inhibición de la apoptosis, con lo que prolongaría la vida de las células41. El trastorno en el bcl2 explicaría en parte la expansión de células B y la gran producción de anticuerpos de estos pacientes, incluyendo el FR y la formación de numerosos inmunocomplejos, algunos de ellos crioprecipitables. Como consecuencia, se desarrollarían distintas enfermedades autoinmunes (hepatitis autoinmune, púrpura trombocitopénica idiopática [PTI], porfiria cutánea tarda, glomerulonefritis, tiroiditis, etc.). La prolongación de la vida de las células B las llevaría a poder sufrir mutaciones genéticas que conducirían al desarrollo de linfomas y otras enfermedades neoplásicas como el carcinoma hepatocelular o el cáncer de tiroides (fig. 7).
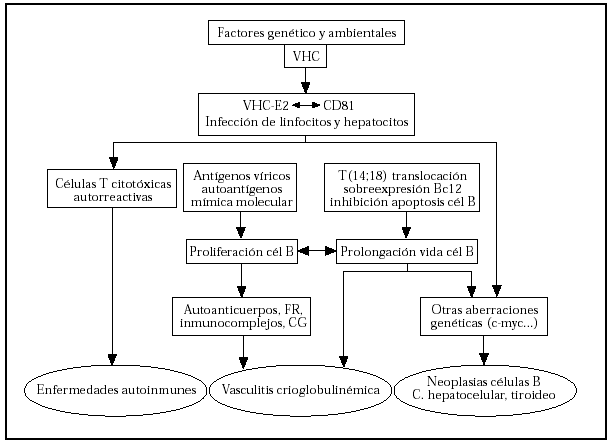
Fig. 7.--Posible etiopatogénesis de la crioglobulinemia mixta y otras enfermedades relacionadas con el virus de la hepatitis C (VHC). FR: factor reumatoide; CG: crioglobulina. Modificada de la cita 1.
Por otra parte, se ha demostrado que las células pluripotenciales CD34+ presentan tanto secuencias del genoma vírico como proteínas. Dado que estas células son capaces de originar todas las líneas celulares sanguíneas, incluyendo los linfocitos B, monocitos y macrófagos, pueden ser el primer sitio de infección y transmitirla posteriormente a su progenia. Además, estas células pluripotenciales se han encontrado en el hígado. Las células pluripotenciales podrían infectarse primeramente en el hígado y desde allí migrar para formar reservorios extrahepáticos en médula ósea o timo, y actuar como células presentadoras de antígeno42.
TRATAMIENTO
El curso clínico, tratamiento y pronóstico en las CGM tipo I dependen de la enfermedad subyacente. Como mencionamos previamente, en casi todos los casos se asocian a enfermedades linfoproliferativas (mieloma múltiple, macroglobulinemia de Waldenström o leucemia linfocítica crónica), por lo que el tratamiento irá dirigido a la enfermedad causante, que suele tratarse con combinaciones de agentes alquilantes (clorambucil o melfalán) y prednisona. Dado que en estos pacientes el síndrome de hiperviscosidad es el hecho principal, la plasmaféresis junto con el tratamiento antes mencionado es la línea de actuación general43.
En el caso de las CGM mixtas tipo II y III podremos tratar la enfermedad desde el punto de vista etiológico, patogénico o sintomático. Es muy importante un correcto estudio del paciente, buscando si existe asociación al VHC, si encontramos alteraciones autoinmunes, y la posibilidad de enfermedades neoplásicas, principalmente linfomas.
Antes del descubrimiento de la asociación entre la CGM y el VHC se utilizaban la prednisona y los fármacos citotóxicos como ciclofosfamida y clorambucil como terapia de mantenimiento en pacientes con enfermedad de lenta evolución, aunque se dudaba si realmente era beneficiosa, salvo en los casos de enfermedad subyacente tratable. Actualmente, la terapia agresiva en el pequeño grupo de pacientes con CGM todavía idiopática se reserva para pacientes con enfermedad severa y de evolución aguda, que se manifiestan con fallo renal, necrosis distal o neuropatía avanzada. En ellos, la plasmaféresis junto con esteroides intravenosos, seguido de prednisona oral y ciclofosfamida es la pauta habitual. También se utiliza la aféresis con criofiltración, que consigue eliminar de manera selectiva las crioproteínas44. No se administrará interferón (IFN) en primera instancia, pues las alteraciones autoinmunes asociadas y los fenómenos de vasculitis pueden exacerbarse.
En los pacientes con CGM mixta asociada al VHC siempre deberemos intentar erradicar el virus, dado que es el factor desencadenante y el responsable de todas las posibles complicaciones autoinmunes y neoplásicas según referimos anteriormente. La eficacia del IFN en el tratamiento de la CGM se conocía antes de que se descubriera su estrecha relación con el VHC. El IFN alfa 2a o alfa 2b han sido la base del tratamiento desde un principio. La pauta más habitual era la de tres millones de unidades tres veces por semana durante un año44, con lo que se conseguían buenos resultados en aproximadamente el 50% de los casos. En ocasiones se asociaba con 16 mg de prednisolona en los 4 días restantes, pero esto no aumentaba el porcentaje de respondedores, aunque sí se conseguía una respuesta más temprana y duradera que con el IFN solo45. Estos efectos se pueden deber a la modulación de los esteroides sobre las quemocinas postinflamatorias inducidas por el IFN-alfa. Actualmente se sabe que la frecuencia de remisiones que consigue la prednisona sola no asociada al IFN es igual a la conseguida con tratamiento placebo, y que los niveles de ARN-VHC aumentan con este tratamiento, lo que parece sugerir que los corticoides favorecen la replicación vírica, al igual que ocurre con otras enfermedades de tipo vírico.
El efecto terapéutico del IFN va ligado directamante a la inhibición de la replicación vírica46. Los niveles de ARN-VHC se tornan indetectables y van seguidos de normalización de las transaminasas y reducción del criocrito. Probablemente se deba a la disminución de antígenos disponibles para la formación de complejos inmunes, con lo que el sistema reticuloendotelial es capaz de un mejor aclaramiento de los mismos. La eficacia del IFN es limitada, sobre todo en determinados genotipos (1a y 1b). Niveles bajos de ARN-VHC predicen una respuesta favorable, mientras que el criocrito y los niveles de FR no son significativos. La regla es la recaída tras unos meses de suspender la terapia. El 80% de los respondedores recaen en los siguientes 6 meses tras cesar el tratamiento. Actualmente se utiliza la asociación de IFN-alfa con ribavirina, un nucleósido con actividad antivírica contra virus ADN y ARN. Existen pocos estudios sobre la eficacia de esta asociación en pacientes con VHC y CGM, pero sí se han constatado sus beneficios en pacientes con infección crónica por VHC47. El porcentaje de respondedores es mayor, el tiempo en conseguir la remisión menor y el período libre de síntomas tras suspender la terapia mayor. El fármaco está contraindicado en pacientes con enfermedad isquémica y/o fallo renal48.
Sin embargo, no todos los síntomas que componen esta heterogénea enfermedad mejoran con el IFN y ribavirina; las úlceras cutáneas, la neuropatía y ciertos casos de fallo renal pueden incluso empeorar con el tratamiento. El potencial antiangiogénico del IFN podría dificultar y retrasar la curación de las lesiones isquémicas.
Una nueva formulación del IFN, el IFN pegilado, parece ofrecer mejores resultados que la fórmula clásica. Una modificación en su molécula consigue grandes cambios en el metabolismo del fármaco, prolonga su vida media y mantiene niveles eficaces en sangre durante más tiempo, lo que permite su administración una sola vez a la semana. Esto reduce la replicación vírica que tenía lugar con el IFN clásico en los días libres de tratamiento49. Además no se pierde en los pacientes sometidos a diálisis.
En los pacientes con CGM mixta asociada a VHC que no respondan al tratamiento anterior y que estén sintomáticos deberemos considerar plasmaféresis, esteroides, dieta pobre en antígenos e inmunosupresores, principalmente ciclofosfamida, para aliviar los síntomas.
En el futuro esperamos el desarrollo de una vacuna contra el virus, o el desarrollo de un bloqueante del receptor para la proteína E2, que impida su unión a la proteína CD81 y evite las numerosas complicaciones autoinmunes y neoplásicas que conlleva. Por último, se espera la aparición del rituximab, un anticuerpo monoclonal dirigido contra el antígeno CD2014.


