







La micosis fungoide (MF) es el linfoma primario cutáneo de células T más frecuente. La evolución clínica clásica de la enfermedad se caracteriza por la progresión desde una fase inespecífica de máculas eritematosas a la aparición de placas y, finalmente, tumores en algunos pacientes. Sin embargo, a lo largo de las últimas décadas se han descrito numerosas variantes de MF, tanto desde el punto de vista clínico como histopatológico, con implicaciones terapéuticas y pronósticas específicas. El diagnóstico diferencial se ve dificultado así ante el amplio abanico de manifestaciones clínicas y patrones histopatológicos de infiltración cutánea, especialmente en fases precoces de la enfermedad. Este artículo revisa las principales características clínicas, histopatológicas e inmunohistoquímicas que definen las distintas variantes de MF descritas en la literatura con el objetivo de facilitar el diagnóstico temprano de MF.
Mycosis fungoides (MF) is the most common primary cutaneous T-cell lymphoma. The clinical course of the disease is typically characterized by progression from a nonspecific phase of erythematous macules to the appearance of plaques and ultimately, in some patients, tumors. However, numerous clinical and histopathologic variants of MF with specific therapeutic and prognostic implications have been described in recent decades. Clarification of the differential diagnosis can be frustrated by the wide range of clinical manifestations and histopathologic patterns of cutaneous infiltration, particularly in the early phases of the disease. In this paper, we review the main clinical, histopathologic, and immunohistochemical characteristics of the variants of MF described in the literature in order to facilitate early diagnosis of the disease.

La micosis fungoide (MF) es el linfoma primario cutáneo de células T más frecuente, representando cerca del 50% del conjunto de todos los linfomas primarios cutáneos1. Descrita por primera vez en 1806 por el dermatólogo francés Jean Louis Alibert, la evolución clásica de la enfermedad se caracteriza por la progresión desde una fase inespecífica de máculas eritematosas de años de evolución a la aparición de placas y, finalmente, tumores en algunos pacientes, cuya morfología característica en forma de seta contribuyó a la denominación de la enfermedad. La presencia de adenopatías, la afectación visceral y la transformación en un linfoma de células grandes constituyen hallazgos menos frecuentes, característicos de estadios avanzados de la enfermedad. A lo largo de las últimas décadas, se han descrito numerosas variantes de MF tanto desde el punto de vista clínico como histopatológico. Aunque algunas de estas variantes constituyen descripciones aisladas de casos, otras presentan mayor relevancia clínica debido a su relativa frecuencia e implicaciones terapéuticas y pronósticas. Así mismo, el amplio abanico de presentaciones clínico-patológicas de la MF en sus fases iniciales hace necesaria su inclusión como parte del diagnóstico diferencial de muchas otras entidades dermatológicas, ya que puede simular gran cantidad de patrones de inflamación cutánea.
El objetivo de este artículo es la revisión de las principales características clínicas, histopatológicas e inmunohistoquímicas que permiten establecer un diagnóstico correcto de MF, especialmente en fases precoces de la enfermedad, así como la descripción de las peculiaridades clínico-patológicas que definen las distintas variantes de MF.
Micosis fungoide clásicaLa definición tradicional de MF contempla la enfermedad como un proceso evolutivo en 3 fases, en función de la aparición progresiva de parches, placas o tumores. Sin embargo, este desarrollo sucesivo no tiene lugar en todos los pacientes, pudiendo permanecer en la fase de placas sin otros signos de progresión de la enfermedad, o presentar tumores asociados a parches y placas como primera manifestación en algunos pacientes.
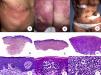
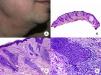
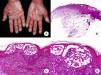
Micosis fungoide en parchesLa MF en parches se caracteriza clínicamente por la presencia de parches y máculas eritematosas, asimétricas e irregulares, asociadas en ocasiones con atrofia y/o telangiectasias2. Se trata de lesiones generalmente asintomáticas o levemente pruriginosas, y transitorias, que desaparecen de forma espontánea sin dejar lesión residual (fig. 1A). Esta fase puede extenderse durante años, en ausencia de otros signos de progresión, y no condiciona un peor pronóstico ni una mayor mortalidad con respecto a la población general3.
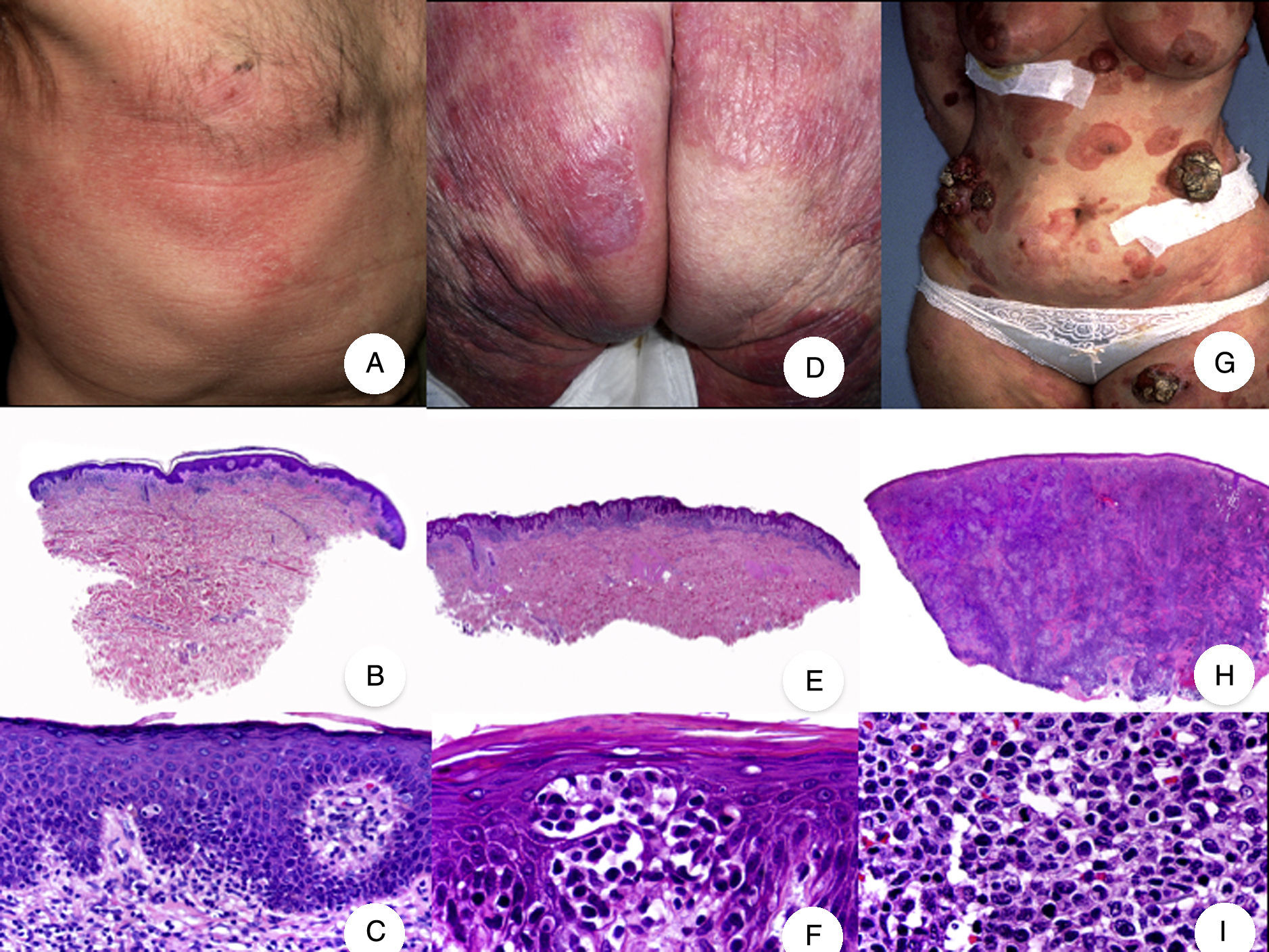
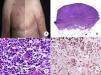
Micosis fungoide clásica. A. Imagen clínica de lesiones de micosis fungoide en fase de parche. B, C. Hallazgos histopatológicos típicos de la fase de parche que muestran infiltrado de linfocitos atípicos en dermis papilar con epidermotropismo. D. Imagen clínica de lesiones de micosis fungoide en fase de placa. E, F. Hallazgos histopatológicos típicos de la fase de placa que muestran hallazgos similares a los de la fase de parche, pero con un infiltrado linfocitario más denso, de distribución liquenoide, dispuesto en banda en la dermis superficial, e intenso epidermotropismo con formación de «microabscesos de Darier-Pautrier». G. Imagen clínica de lesiones de micosis fungoide en fase tumoral. H, I. Hallazgos histopatológicos típicos de la fase tumoral que muestran un infiltrado linfocitario difuso compuesto por linfocitos grandes y pleomórficos que ocupa todo el espesor de la dermis.
Los hallazgos histopatológicos de la MF en parches se caracterizan por la presencia de escasos linfocitos dispersos en las capas basales de la epidermis, acompañados de paraqueratosis focal, fibrosis de la dermis papilar y mayor número de linfocitos dispuestos en banda a lo largo de la unión dermoepidérmica. Así mismo, es posible observar la presencia de un infiltrado predominantemente linfocitario, de disposición perivascular, perianexial o subepidérmico, con presencia de eosinófilos y algunas células plasmáticas (fig. 1 B y C). Los linfocitos intraepidérmicos son generalmente más pleomórficos y de mayor tamaño que los de la dermis superficial, y en ocasiones presentan un halo citoplasmático claro. Estos hallazgos morfológicos son altamente indicativos de MF, pero no son constantes, y a menudo se requiere el análisis de biopsias cutáneas sucesivas a lo largo del tiempo para establecer el diagnóstico definitivo de MF en fase de parches4.
Micosis fungoide en placasLos pacientes con MF en placas se caracterizan clínicamente por la presencia de placas bien delimitadas y frecuentemente pruriginosas, eritematosas o de color parduzco, infiltradas al tacto y, a menudo, asociadas a descamación, que afectan generalmente a una amplia superficie cutánea (fig. 1D). Estas lesiones coexisten frecuentemente con máculas características de fases más precoces de MF, ya sea de forma contigua o en otras áreas de la piel5,6.
Desde el punto de vista histopatológico, la MF en placas se caracteriza por hallazgos similares a los de la fase de parches, pero con un infiltrado linfocitario más denso, de distribución liquenoide, dispuesto en banda en la dermis superficial. Este infiltrado se compone principalmente de linfocitos de tamaño pequeño y mediano, un núcleo hipercromático y pleomórfico, y numerosas circunvoluciones, que le confieren un aspecto «cerebriforme» (fig. 1 E y F). También es frecuente observar un mayor epidermotropismo de este infiltrado que en las lesiones en parche, caracterizado por la presencia de linfocitos atípicos en la epidermis, ya sea de manera aislada o formando colecciones que reciben el nombre de «microabscesos de Darier-Pautrier». Otros hallazgos frecuentes incluyen la hiperplasia epidérmica, la fibrosis de la dermis papilar, así como la presencia de algunos eosinófilos y células plasmáticas en el infiltrado inflamatorio acompañante.
Micosis fungoide tumoralDesde el punto de vista clínico, el hallazgo fundamental de la MF tumoral es la presencia de tumores cutáneos, a menudo coexistentes con parches y placas (fig. 1G). La ausencia de estas últimas debe replantear el diagnóstico de MF, y establecer el diagnóstico diferencial con otros linfomas cutáneos no MF más agresivos. Los tumores se caracterizan por un importante crecimiento vertical, dando lugar a la formación de nódulos de superficie lisa, coloración marrón rojiza o rojo azulada, y que en ocasiones pueden alcanzar un tamaño de varios centímetros, ulcerarse en la superficie y sobreinfectarse7.
Histopatológicamente se observa un infiltrado linfocitario difuso que adopta una morfología nodular, compuesto por linfocitos grandes y pleomórficos con núcleo hipercromático y nucléolo prominente, que ocupan todo el espesor de la dermis y pueden extenderse al tejido celular subcutáneo (fig. 1 H e I). Es frecuente observar además un elevado índice proliferativo del infiltrado, con abundantes figuras de mitosis tanto típicas como atípicas. Los microabscesos de Darier-Pautrier, al igual que el epidermotropismo, se encuentran normalmente ausentes en esta fase.
Diagnóstico de micosis fungoideEl diagnóstico de MF en fases iniciales constituye un reto para el dermatopatólogo, no solo por la sutileza de los hallazgos histopatológicos, sino también por el solapamiento con otras dermatosis inflamatorias. Además, en las fases tempranas de la enfermedad es frecuente la ausencia de algunos de los rasgos clínicos e histopatológicos característicos de MF. De esta forma, los microabscesos de Darier-Pautrier se observan en menos del 25% de los casos de MF en fases iniciales, los linfocitos atípicos están presentes en menos del 10% de las biopsias de estos pacientes, y el epidermotropismo, que constituye un rasgo característico de MF, puede estar ausente en hasta un 4% de los casos1,8.
Por esta razón, la Sociedad Internacional de Linfomas Cutáneos –International Society for Cutaneous Lymphomas– desarrolló en 2005 un algoritmo diagnóstico basado en una combinación de criterios clínicos, histopatológicos, inmunohistoquímicos y moleculares con el objetivo de facilitar el diagnóstico de MF en estadios iniciales (tabla 1)9, que apoya la utilidad de las biopsias cutáneas seriadas en aquellos casos con un diagnóstico ambiguo10.
Algoritmo para el diagnóstico de micosis fungoide en estadios iniciales
| Criterios | Puntuación |
|---|---|
| Clínica Criterio principal Parches y placas persistentes y/o progresivos Criterios adicionales Áreas no fotoexpuestas Variación en tamaño y forma Poiquilodermia | 2 puntos si criterio principal y 2 criterios adicionales 1 punto si criterio principal y un criterio adicional |
| Histopatología Criterio principal Infiltrado linfocítico superficial Criterios adicionales Epidermotropismo sin espongiosis Atipia linfocítica (células con núcleo grande, hipercromático e irregular o «cerebriforme») | 2 puntos si criterio principal y 2 criterios adicionales 1 punto si criterio principal y un criterio adicional |
| Biología molecular Reordenamiento monoclonal del gen γTCR | 1 punto si clonalidad |
| Inmunopatología <50% de células T CD2+, CD3+ y/o CD5 <10% de células T CD7+ Discordancia dermoepidérmica de CD2, CD3, CD5 o CD7a | 1 punto si al menos un criterio |
Son necesarios al menos 4 puntos para el diagnóstico de micosis fungoide, independientemente de la combinación de criterios clínicos, histopatológicos, biomoleculares e inmunohistoquímicos.
Pérdida de expresión de antígenos de células T limitada a la epidermis.
Fuente: Adaptada de Pimpinelli et al9.
El análisis inmunohistoquímico de las células T neoplásicas contribuye, junto con los criterios clínicos e histopatológicos, al diagnóstico de MF. Los linfocitos característicos de la MF son habitualmente CD4+, presentando pérdida variable en la expresión de otros marcadores de células T, como CD2, CD3, CD5, CD7 y CD26. De los anteriores, la pérdida de expresión de CD7 constituye el hallazgo más frecuentemente observado, seguido por la pérdida de expresión de CD5. De todas formas, en hasta un 20% de los casos es posible encontrar un fenotipo CD8+. Por otra parte, la presencia de un reordenamiento monoclonal del gen γ del receptor de células T constituye un criterio adicional para el diagnóstico de MF en fases tempranas. Desafortunadamente, tanto la pérdida de expresión de CD7 como la presencia de un reordenamiento monoclonal del gen γ del receptor de células T no permiten un diagnóstico inequívoco de MF, al tratarse de hallazgos inespecíficos que pueden observarse también en otras dermatosis inflamatorias benignas, debido probablemente a la estimulación crónica de los linfocitos T11.
A pesar de la ausencia de marcadores celulares y moleculares que permitan un diagnóstico de certeza de MF en fases tempranas, estudios recientes han identificado hasta 19 genes distintos que muestran un aumento diferencial de su expresión en lesiones precoces de MF con respecto a otras dermatosis inflamatorias crónicas12. Entre estos genes destacan especialmente programmed cell death protein 1 (PDCD1) y thymocyte selection-associated high mobility group box protein (TOX), por su capacidad de discriminación entre ambos procesos. En concreto, TOX parece constituir un marcador inmunohistoquímico sensible y específico para el diagnóstico precoz de MF, y muestra un patrón de tinción nuclear intensa y difusa, ausente en otras dermatosis inflamatorias. Las células que muestran positividad para TOX se caracterizan por la presencia de núcleos atípicos, y se observan tanto en la epidermis como en la dermis papilar de biopsias cutáneas de MF en estadios iniciales, así como en las células que constituyen los microabscesos de Darier-Pautrier. De hecho, se ha evidenciado una estrecha correlación entre la detección de TOX y las células tumorales de la MF12.
Variantes clínico-patológicas de micosis fungoideEl espectro de variantes clínico-patológicas de MF incluye variantes clínicas, definidas por la presentación clínica con hallazgos histopatológicos similares a la MF clásica, variantes clínico-patológicas, con peculiaridades tanto clínicas como histopatológicas, y variantes histopatológicas, distinguibles de la MF clásica exclusivamente mediante el análisis de la biopsia (tabla 2). Del conjunto de estas variantes, solo la MF foliculotropa, la reticulosis pagetoide y el síndrome de la piel laxa granulomatosa están incluidas en la última clasificación internacional de los linfomas cutáneos elaborada por la Organización Mundial de la Salud (OMS) en 2016. Sin embargo, el resto de las variantes de MF descritas en esta revisión han sido incluidas por la importancia de su conocimiento por dermatólogos y dermatopatólogos, y para ayudar así en el diagnóstico diferencial de esta enfermedad.
Variantes clínicas, clínico-patológicas e histológicas de la micosis fungoide
| Variantes clínicas |
|---|
| MF hipopigmentada MF eritrodérmica MF ictiosiforme MF palmar y plantar MF papilomatosa MF papular MF solitaria o unilesional MF invisible |
| Variantes clínico-patológicas |
|---|
| MF foliculotropa MF con quistes infundibulares eruptivos MF siringotrópica Síndrome de la piel laxa granulomatosa Reticulosis pagetoide o enfermedad de Woringer-Kolopp MF poiquilodérmica (poiquilodermia atrófica vascular) MF ampollosa y MF dishidrótica MF anetodérmica MF hiperpigmentada MF purpúrica MF pustulosa MF verrucosa |
| Variantes histopatológicas |
|---|
| MF granulomatosa MF intersticial MF con trasformación en células grandes |
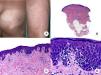
La MF hipopigmentada es una variante clínica poco frecuente de MF que clínicamente se caracteriza por la presencia de máculas y placas hipopigmentadas con ausencia de atrofia cutánea (fig. 2A). Se observa con mayor frecuencia en pacientes con fototipos oscuros y constituye una de las variantes más frecuentes durante la edad pediátrica, aunque también se ha descrito su aparición en pacientes adultos13. Presenta mejor pronóstico que la MF clásica, y responde de forma adecuada al tratamiento con fototerapia UVB de banda estrecha, especialmente en niños14, produciéndose una repigmentación gradual de las lesiones tras el tratamiento. Sin embargo, aunque en ocasiones las lesiones hipopigmentadas constituyen la única manifestación de la enfermedad, una exploración física cuidadosa a menudo permite detectar la presencia de parches o placas eritematosas clásicas coexistentes.
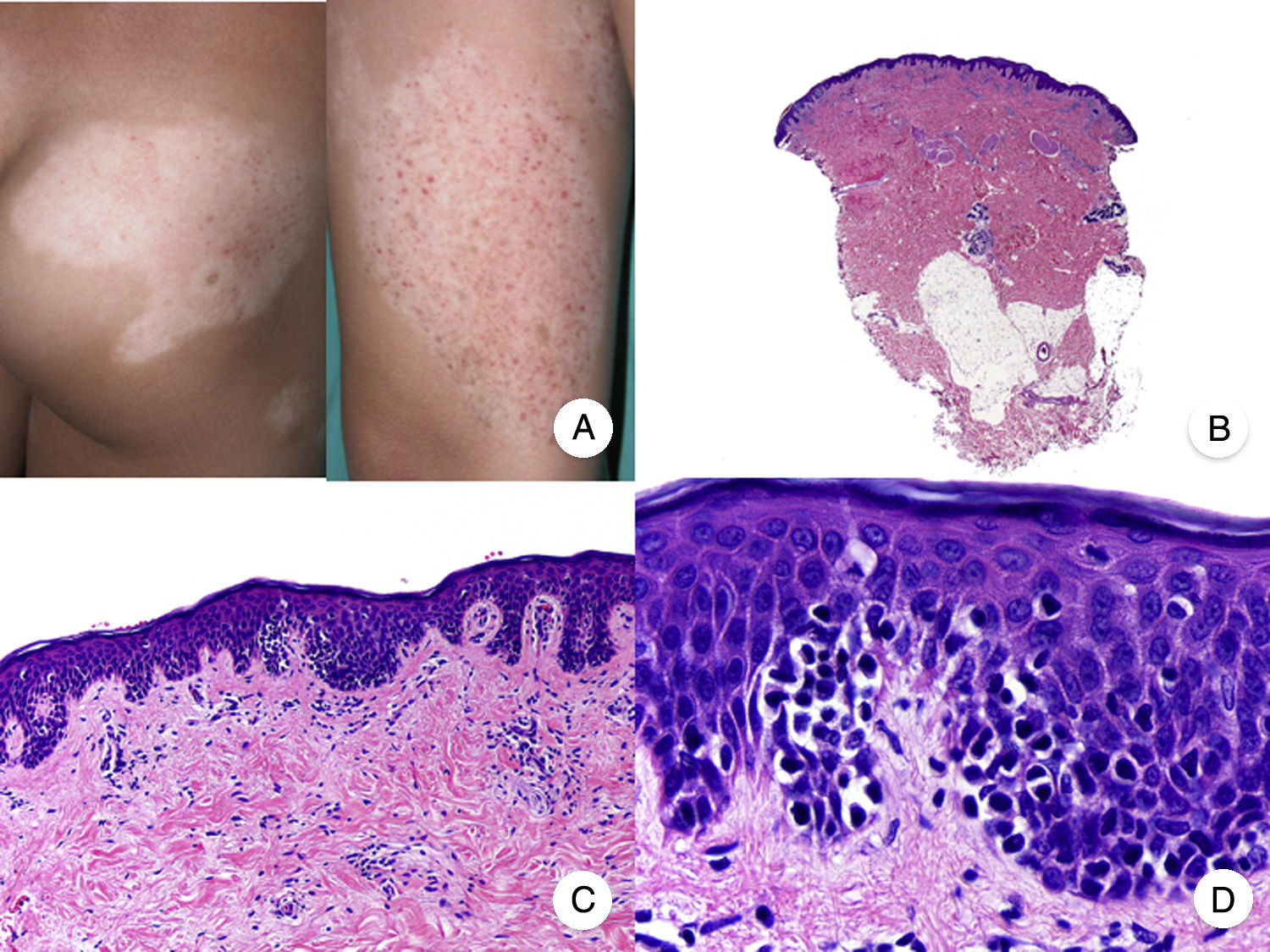
Micosis fungoide hipopigmentada. A. Imágenes clínicas que muestran máculas y placas hipopigmentadas con ausencia de atrofia cutánea en un niño. B. Vista panorámica que muestra un infiltrado a nivel de la dermis superficial. C, D. Detalle del infiltrado dérmico con linfocitos atípicos y epidermotropismo.
Desde el punto de vista histopatológico, es frecuente observar los hallazgos típicos de la MF clásica, con un llamativo epidermotropismo constituido por linfocitos atípicos, a menudo CD8+. Otros hallazgos más inespecíficos que también se observan en ocasiones son un infiltrado linfocitario dérmico perianexial y perivascular, e hiperplasia epidérmica de patrón psoriasiforme (fig. 2 B-D). El patrón inmunofenotípico característico de esta variante de MF muestra una disminución de la expresión de CD7, así como una mayor presencia epidérmica de células de Langerhans CD1a+, habiéndose descrito también casos con un patrón CD4+. La expresión de CD30 es excepcional13,15.
El principal diagnóstico diferencial debe realizarse con las lesiones de vitíligo inflamatorio16,17, que muestran también un epidermotropismo linfocitario CD8+, siendo a menudo necesarios la correlación clínico-patológica y el seguimiento del paciente para poder diferenciar ambas entidades. También es importante incluir en el diagnóstico diferencial otras dermatosis que cursan con hipopigmentación, como la pitiriasis alba.
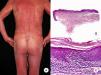
Micosis fungoide eritrodérmicaEl término MF eritrodérmica hace referencia al desarrollo de eritrodermia en pacientes con hallazgos histopatológicos característicos de MF clásica, en ausencia de criterios diagnósticos de síndrome de Sézary (fig. 3)18. La aparición de eritrodermia puede constituir raramente la primera manifestación de la enfermedad o presentarse más frecuentemente tras el desarrollo de parches y placas típicos de MF, a menudo en relación con tratamientos inadecuados, facilitando así el diagnóstico de la misma19. Los pacientes con MF eritrodérmica desarrollan nuevamente lesiones de MF clásica tras el tratamiento apropiado de la eritrodermia.
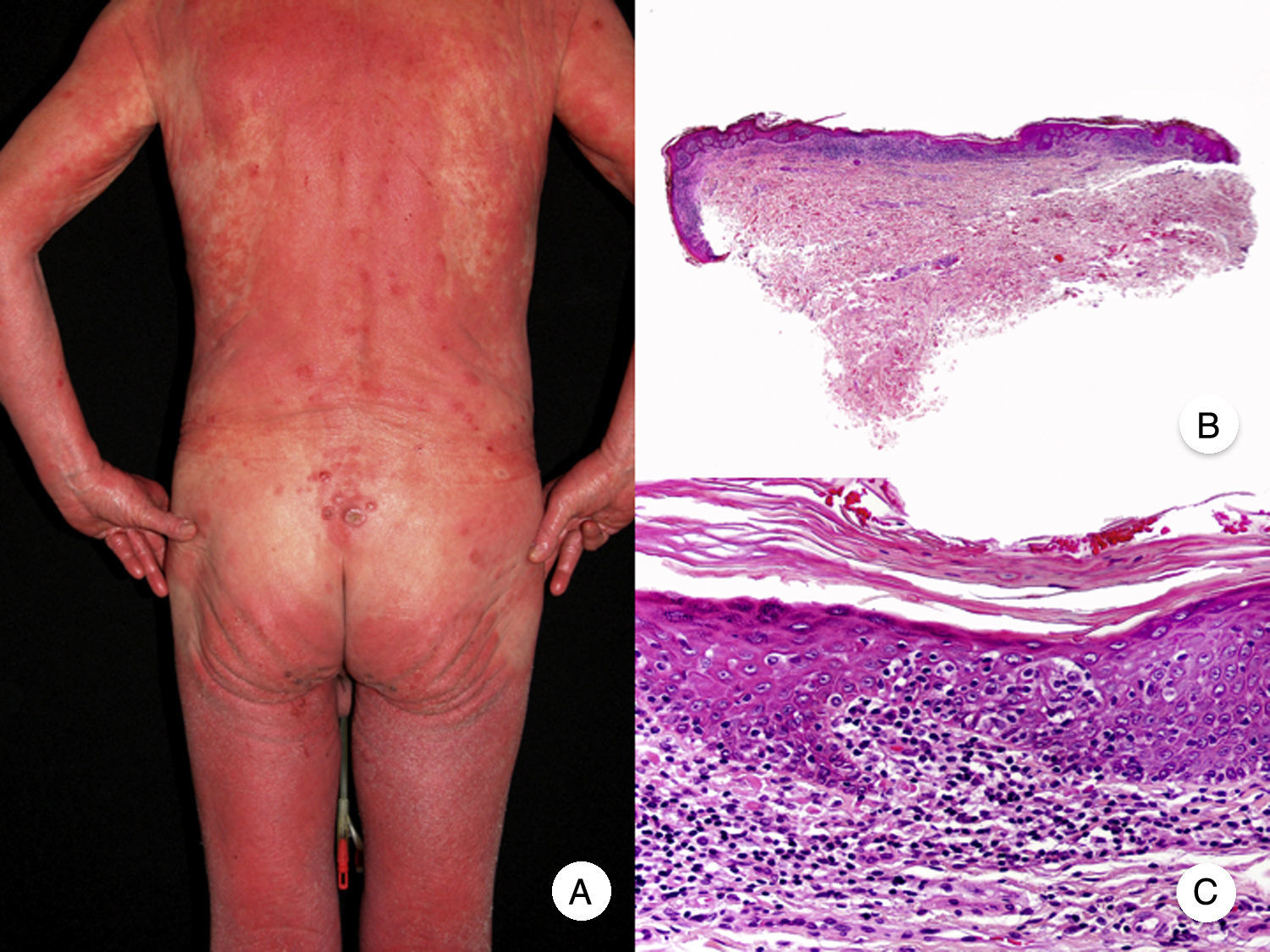
El diagnóstico diferencial con el síndrome de Sézary se basa en la ausencia de hallazgos hematológicos propios de este último, siendo posible, aunque infrecuente, la presencia de linfadenopatías. Aunque ambos procesos comparten estadificación y clasificación desde el punto de vista hematooncológico20,21, estudios recientes evidencian alteraciones cromosómicas22 y expresión inmunofenotípica23 diferentes, que demuestran un origen en distintas poblaciones linfocitarias, constituyendo, por lo tanto, 2 entidades independientes.
Micosis fungoide ictiosiformeLa MF ictiosiforme es una variante clínica de MF muy poco frecuente. Se caracteriza por la presencia de placas geométricas con un patrón adoquinado que recuerda clínicamente a la ictiosis vulgar, y afecta generalmente a las extremidades24. En ocasiones se asocia a pápulas foliculares y otras lesiones características de la MF foliculotropa.
Desde el punto de vista histopatológico, los hallazgos son similares a aquellos observados en la MF clásica, junto con otros característicos de ictiosis, como hipogranulosis e hiperqueratosis25. La coexistencia de datos de ambas entidades en la misma biopsia permite realizar el diagnóstico diferencial con la ictiosis paraneoplásica en un paciente con MF conocida, donde solo se observan las alteraciones propias de la ictiosis en ausencia de la infiltración linfocitaria característica de MF. La infiltración del folículo piloso por linfocitos atípicos, observada tanto en ausencia como en presencia de pápulas foliculares, aunque no de forma constante, parece indicar una asociación más estrecha con la MF foliculotropa25.
Micosis fungoide palmar y plantarLa MF palmar y plantar es una variante clínica poco frecuente de MF, caracterizada por la afectación exclusiva de palmas y plantas en forma de parches, placas o tumores26. Las lesiones pueden extenderse al dorso de manos y pies, dedos y muñecas, siendo frecuente la presencia de onicodistrofia. La aparición de lesiones típicas de MF en zonas acras debe ir acompañada de ausencia de las mismas en cualquier otra localización corporal. Esta variante presenta un curso clínico indolente, siendo inusual el compromiso de otras áreas de la piel27.
Los hallazgos histológicos de la MF palmar y plantar son similares a los observados en la MF clásica, en contraste con los linfocitos atípicos de aspecto pagetoide y el intenso epidermotropismo de la enfermedad de Woringer-Kolopp.
Micosis fungoide papilomatosaLa MF papilomatosa o vegetante se caracteriza por la presencia de lesiones que recuerdan a la acantosis nigricans o a queratosis seborreicas, localizadas generalmente en áreas flexurales como cuello, axilas y pliegues inguinales. Histopatológicamente se observa una marcada acantosis y papilomatosis junto con un infiltrado difuso en banda, compuesto por linfocitos atípicos, y con presencia de epidermotropismo28,29.
Micosis fungoide papularLa MF papular es una variante clínica de MF cuya expresión morfológica característica son pápulas de pequeño tamaño no foliculocéntricas, en ausencia de parches y placas clásicas de MF (fig. 4A)30. Los hallazgos histopatológicos observados son similares a los de la MF clásica, característicamente de forma parcheada y sin presencia de afectación folicular (fig. 4 B-D). Aunque inicialmente se describió como una variante de buen pronóstico, con un comportamiento benigno a largo plazo31, también se han observado casos con una rápida progresión a fase tumoral o eritrodermia en cortos periodos de tiempo32.
El principal reto en el reconocimiento de esta variante radica en el diagnóstico diferencial con la papulosis linfomatoide y otros trastornos linfoproliferativos primarios cutáneos, en ausencia de manifestaciones cutáneas típicas de MF clásica. La evolución de las lesiones en la MF papular tiende a ser estable, no produciéndose fenómenos de regresión espontánea ni ulceración, hemorragia o cicatriz residual varioliforme, observados con frecuencia en la papulosis linfomatoide. Así mismo, el inmunofenotipo del infiltrado en la MF papular es habitualmente CD30−33.
Micosis fungoide solitaria o unilesionalSe define como MF solitaria o unilesional a la aparición aislada de una mácula, placa o nódulo con características histopatológicas indistinguibles de la MF clásica, sin evidencia de otras lesiones cutáneas indicativas de MF34,35. La presencia de un infiltrado inflamatorio en banda acompañado de epidermotropismo de linfocitos atípicos aislados, sin otros hallazgos histopatológicos acompañantes, orienta en el diagnóstico diferencial con la reticulosis pagetoide localizada, otra variante de MF que se manifiesta en forma de lesiones aisladas pero con características clínicas e histológicas diferenciales36. La MF solitaria o unilesional es una variante de buen pronóstico, con un comportamiento biológico benigno, que raramente evoluciona a formas avanzadas de la enfermedad.
Micosis fungoide invisibleEl término MF invisible hace referencia a la presencia de hallazgos histopatológicos característicos de MF en piel de aspecto normal, sin lesiones cutáneas visibles, siendo el prurito el único síntoma. La ausencia de lesiones cutáneas puede persistir a lo largo de todo el curso clínico de la enfermedad37.
Variantes clínico-patológicas de micosis fungoideMicosis fungoide foliculotropaLa MF foliculotropa constituye una de las variantes más frecuentes de MF, alcanzando hasta el 10% de los pacientes en algunas series38, y muestra una identidad propia, suficiente como para haber sido incluida en la clasificación de linfomas cutáneos elaborada en colaboración entre la OMS y la Organización Europea para la Investigación y el Tratamiento del Cáncer20, así como en la Clasificación de los tumores hematopoyéticos y de los tejidos linfoides publicada por la OMS en 201621.
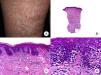
La presentación clínica de la MF foliculotropa suele ser en forma de placas induradas, eritematosas, asociadas con pápulas foliculares y lesiones acneiformes, con la presencia de pequeños quistes y comedones, que se localizan a menudo en la cabeza y el cuello, y frecuentemente asocian alopecia y ocasionalmente mucinorrea (fig. 5A). Además, la presencia de placas infiltradas con alopecia de cejas constituye un hallazgo característico. Esta variante de MF se asocia generalmente con un importante prurito, que suele ser refractario al tratamiento y conlleva un mal pronóstico, similar al de la fase tumoral de la MF clásica39,40. La variante foliculotropa de MF afecta principalmente a adultos, con predominio del sexo masculino, aunque también se han descrito casos en la infancia y la adolescencia20.
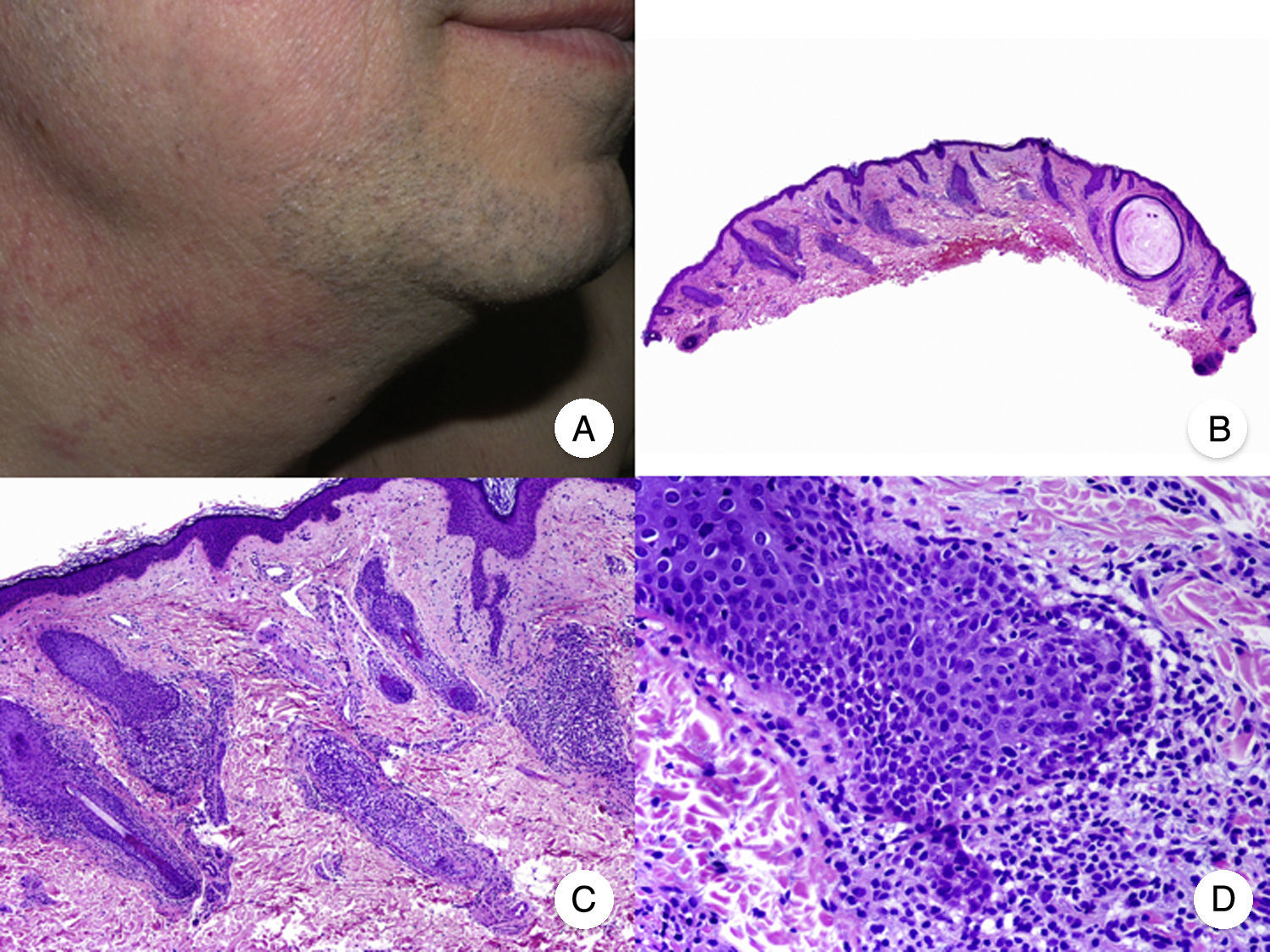
Micosis fungoide foliculotropa. A. Imagen clínica que muestra pápulas foliculares y lesiones acneiformes en la cara y el cuello. B. Vista panorámica que muestra un infiltrado rodeando los folículos pilosos. C, D. A mayor aumento se observan linfocitos atípicos agrupados en el seno del epitelio de la vaina radicular externa del folículo piloso.
Desde el punto de vista histopatológico, la MF foliculotropa se caracteriza por la presencia de linfocitos atípicos aislados o agrupados en el seno del epitelio de la vaina radicular externa del folículo piloso, fenómeno conocido como «foliculotropismo», junto con un infiltrado inflamatorio dérmico perivascular y perianexial, compuesto por linfocitos atípicos, eosinófilos y células plasmáticas (fig. 5 B-D). Aunque es posible observar la presencia de linfocitos aislados salpicando la epidermis, el epidermotropismo no es un hallazgo frecuente en esta variante de MF41. En algunos casos, es posible observar además un importante infiltrado inflamatorio afectando a las glándulas sudoríparas ecrinas, hecho que orienta hacia una afectación anexial generalizada. Finalmente, en muchas ocasiones se observa degeneración mucinosa del epitelio folicular (mucinosis folicular), que se hace más evidente mediante la tinción con azul Alcian o cualquier otra tinción de mucina mesenquimal. La inmunohistoquímica es similar a la de la MF clásica, siendo frecuente la presencia de un infiltrado asociado de células CD30+20.
Micosis fungoide con quistes infundibulares eruptivosLa aparición de una erupción folicular localizada o generalizada en forma de quistes infundibulares y comedones, descrita en algunos pacientes con MF, parece constituir una manifestación rara de la enfermedad42–44. En ocasiones, el tamaño y la inflamación de las lesiones puede simular la MF tumoral45. Clínicamente debe realizarse el diagnóstico diferencial con la variante foliculotropa, si bien algunos autores consideran ambas variantes como parte del mismo espectro de afectación folicular dentro de la enfermedad32.
Histopatológicamente, se observan los hallazgos característicos de un quiste infundibular rodeado por un denso infiltrado, compuesto predominantemente por linfocitos atípicos que afectan a la pared del quiste. La aparición de quistes infundibulares puede estar en relación con la afectación específica del orificio folicular por linfocitos neoplásicos, condicionando el taponamiento y la dilatación posterior del infundíbulo42.
Micosis fungoide siringotrópicaLa MF siringotrópica es una variante clínico-patológica poco frecuente de MF, caracterizada por la afectación de las glándulas ecrinas. Clínicamente se presenta en forma de pápulas y placas eritematosas, en ocasiones hiperpigmentadas, que pueden ir acompañadas de una erupción folicular. Además, como consecuencia de la afectación anexial, es frecuente la aparición de anhidrosis y alopecia46. Las lesiones pueden ser únicas o generalizadas, asociando en ocasiones lesiones típicas de MF clásica en otras áreas de la piel, y siendo frecuente la afectación de palmas y plantas. Se trata de una variante de MF con un curso clínico indolente.
La afectación de las glándulas y los conductos ecrinos por un infiltrado denso de linfocitos atípicos y la presencia de un grado variable de siringometaplasia (transformación escamosa del epitelio glandular) constituye el dato histopatológico más característico, permitiendo el diagnóstico diferencial con la MF foliculotropa (fig. 6)47. Así mismo, a menudo se observa la presencia de epidermotropismo y afectación folicular46. Otros hallazgos menos específicos incluyen la existencia de un infiltrado inflamatorio en banda e hiperplasia epidérmica. El cuadro clásicamente denominado hiperplasia siringolinfoide se considera en la actualidad que corresponde a una MF siringotrópica.
Micosis fungoide siringotrópica. A. Vista panorámica que muestra un infiltrado en banda en dermis reticular y periglandular. B. A mayor aumento se observa la afectación de las glándulas ecrinas por un infiltrado denso de linfocitos. C, D. A mayor aumento se observan linfocitos atípicos rodeando una glándula ecrina con siringometaplasia.
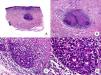
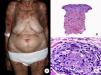
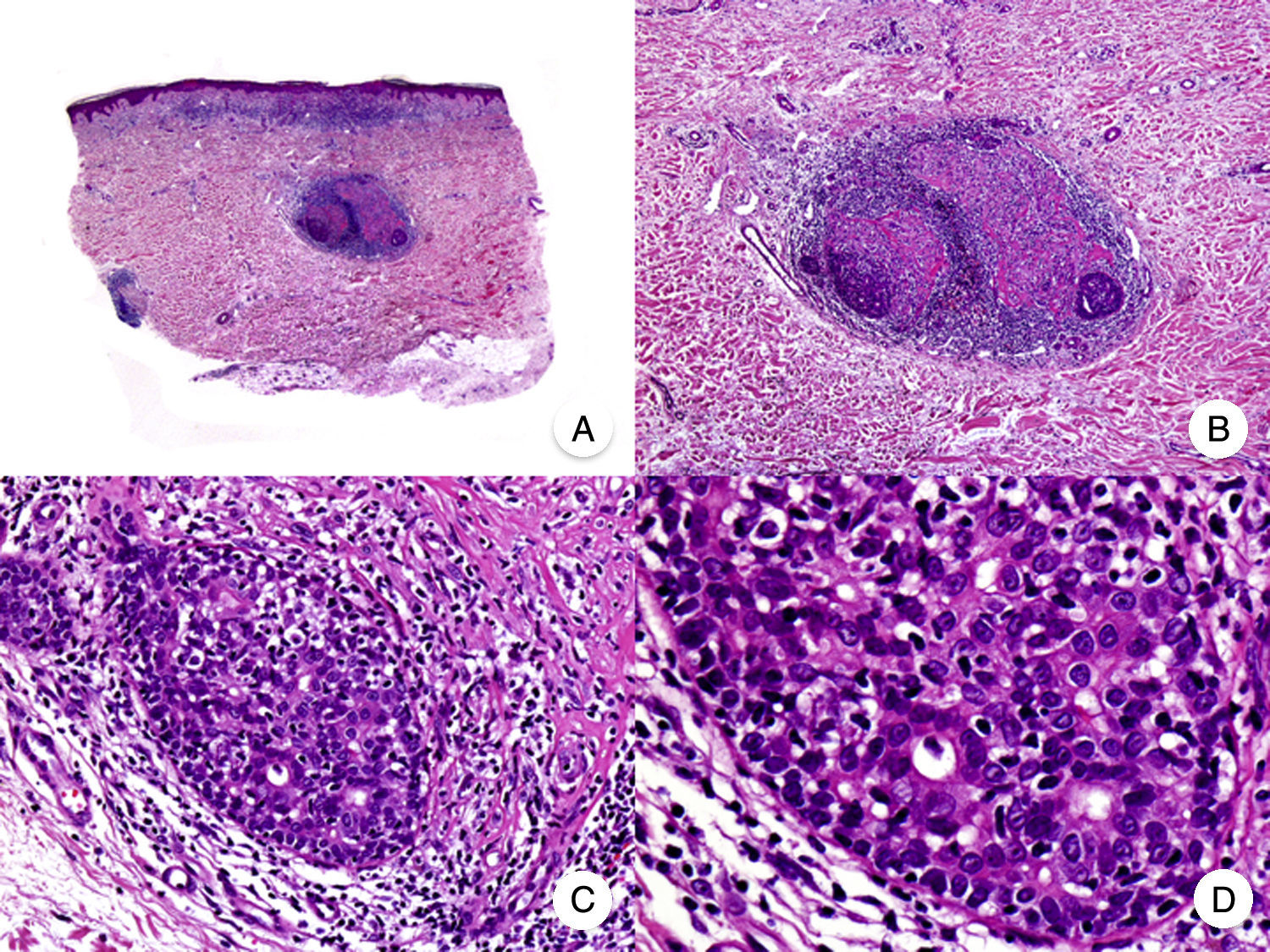
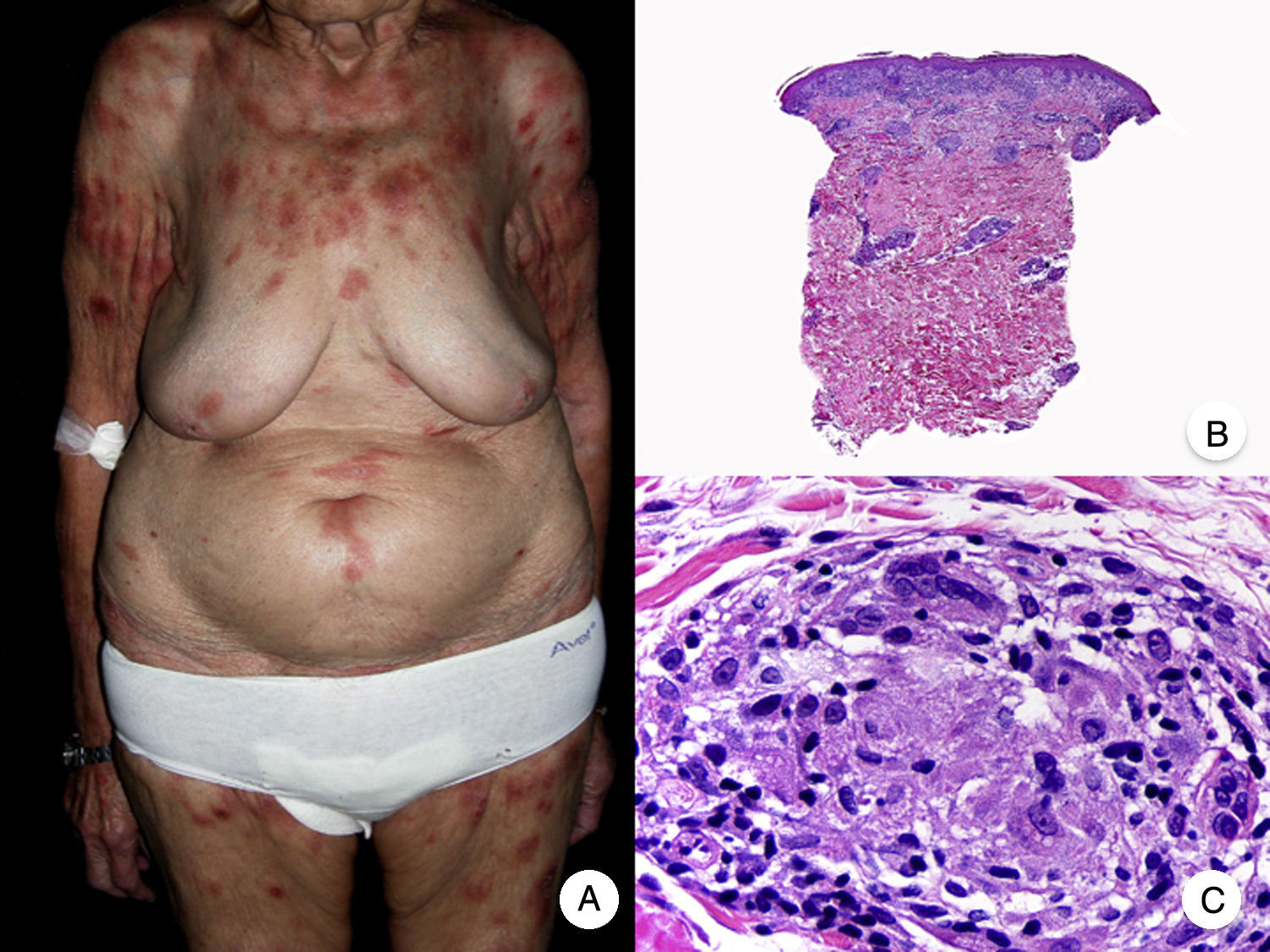
El síndrome de la piel laxa granulomatosa es una variante muy poco frecuente de MF, caracterizada por la presencia de placas eritematosas en forma de pliegues colgantes de piel laxa, a menudo voluminosos, localizados generalmente en áreas flexurales como axilas e ingles (fig. 7A)48. La llamativa expresión clínica, junto con los hallazgos histopatológicos característicos, han contribuido a su inclusión en la última clasificación de linfomas cutáneos elaborada por la OMS y la Organización Europea para la Investigación y el Tratamiento del Cáncer20,21. Se trata de una variante de MF con un comportamiento biológico indolente y tendencia a la recidiva tras la extirpación local, afectando con mayor frecuencia a pacientes jóvenes. Así mismo, estos pacientes presentan con frecuencia un segundo linfoma, sobre todo enfermedad de Hodgkin20.
Síndrome de la piel laxa granulomatosa. A. Imagen clínica que muestra una placa eritematosa en forma de pliegue colgante voluminoso, en la axila. B. Vista panorámica que muestra un infiltrado inflamatorio difuso afectando al espesor completo de la dermis y extendiéndose al tejido celular subcutáneo. C. A mayor detalle se observa que el infiltrado está compuesto por histiocitos y linfocitos atípicos, así como células gigantes multinucleadas con gran cantidad de núcleos.
Los hallazgos histopatológicos característicos de esta entidad incluyen la presencia de un infiltrado inflamatorio difuso de carácter granulomatoso, afectando al espesor completo de la dermis y, en ocasiones, extendiéndose al tejido celular subcutáneo. Este infiltrado está compuesto por histiocitos y linfocitos atípicos, así como células gigantes multinucleadas, que a menudo contienen gran cantidad de núcleos. Además, es característico observar una importante pérdida –o incluso ausencia completa– de fibras elásticas, pudiéndose observar fragmentos de las mismas en el interior de las células gigantes multinucleadas, fenómeno conocido como elastofagocitosis (fig. 7 B y C). También es frecuente observar linfocitos incorporados mediante fagocitosis o emperipolesis en el interior de las células gigantes multinucleadas, siendo llamativa la ausencia de epidermotropismo y microabscesos de Darier-Pautrier. En contraste con estos hallazgos, en la MF granulomatosa suelen observarse pequeños granulomas sarcoideos salpicados por la dermis y ausencia de elastofagocitosis. Sin embargo, el diagnóstico diferencial entre ambas variantes se basa fundamentalmente en las manifestaciones clínicas49.
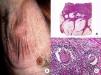
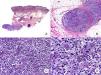
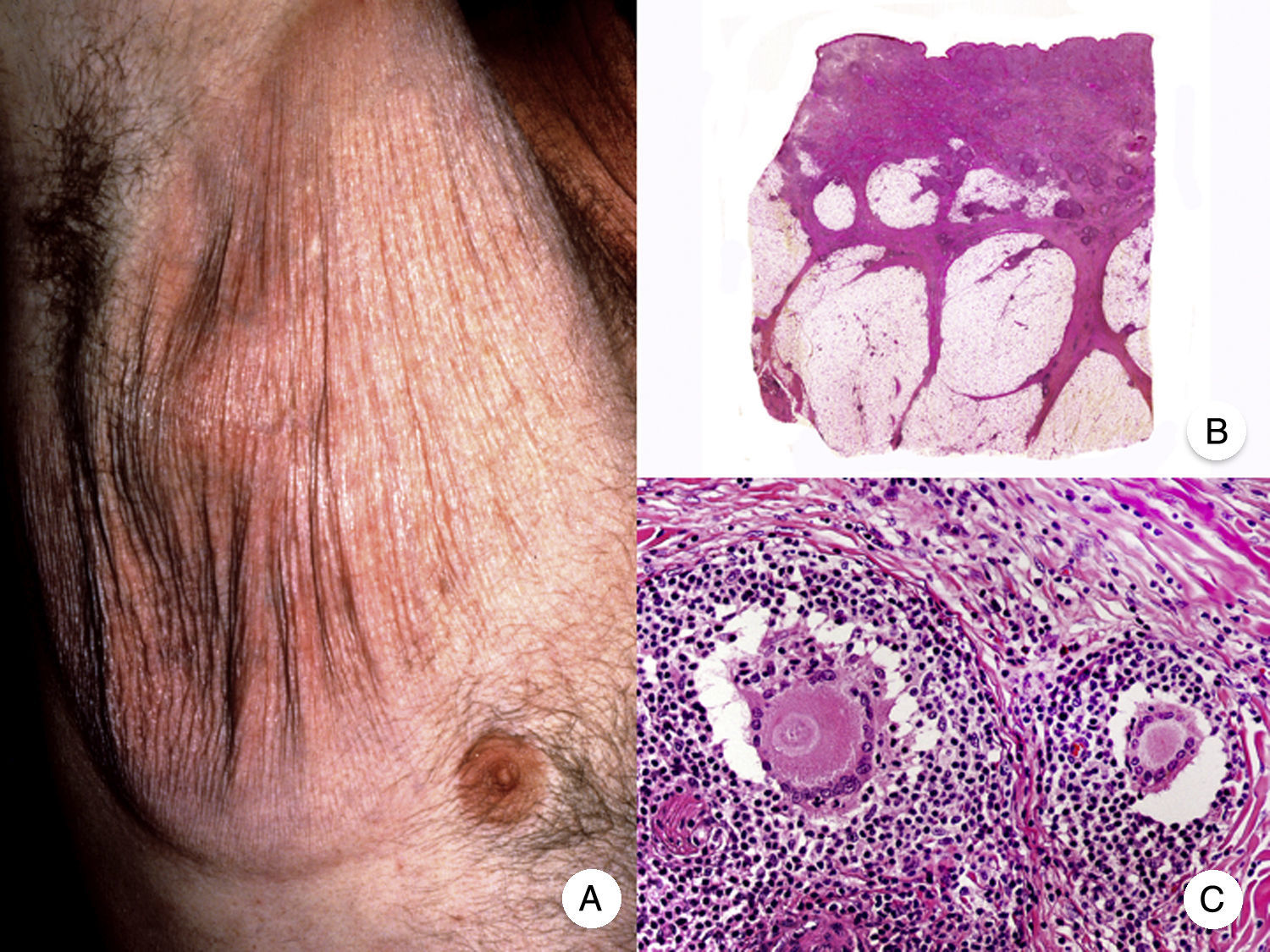
Reticulosis pagetoideLa reticulosis pagetoide o enfermedad de Woringer-Kolopp es una variante rara de MF de crecimiento lento, curso indolente y muy buen pronóstico, incluida en la última clasificación de linfomas cutáneos elaborada por la OMS y la Organización Europea para la Investigación y el Tratamiento del Cáncer20,21. Clínicamente se caracteriza por la aparición de una lesión aislada, localizada generalmente en zonas acras de las extremidades. Suele ser una lesión en forma de placa, de aspecto psoriasiforme, hiperqueratósica y eritematosa (fig. 8A). El tratamiento de elección es la escisión quirúrgica o la radioterapia local. La presencia de progresión o afectación extracutánea, clásicamente denominada forma diseminada o enfermedad de Ketron-Goodman, se considera actualmente que corresponde a otros linfomas cutáneos más agresivos, casi siempre a un linfoma T citotóxico epidermotropo agresivo20.
Reticulosis pagetoide. A. Imagen clínica que muestra una placa única, de aspecto psoriasiforme, hiperqueratósica y eritematosa, en el talón. B. Vista panorámica que muestra hiperplasia epidérmica, hiperqueratosis con paraqueratosis y un infiltrado en dermis papilar. C, D. A mayor detalle se puede observar el marcado epidermotropismo del infiltrado, compuesto por linfocitos atípicos de aspecto pagetoide, con un núcleo grande e hipercromático.
Los hallazgos histopatológicos característicos de la reticulosis pagetoide incluyen un marcado epidermotropismo del infiltrado, compuesto por linfocitos atípicos de aspecto pagetoide, con un núcleo grande e hipercromático rodeado de un halo claro, e hiperplasia epidérmica con hiperqueratosis y paraqueratosis (fig. 8 B-D). Esta variante de MF presenta a menudo un inmunofenotipo CD8+, expresando casi siempre CD3050, sin que ello implique un comportamiento biológico más agresivo51.
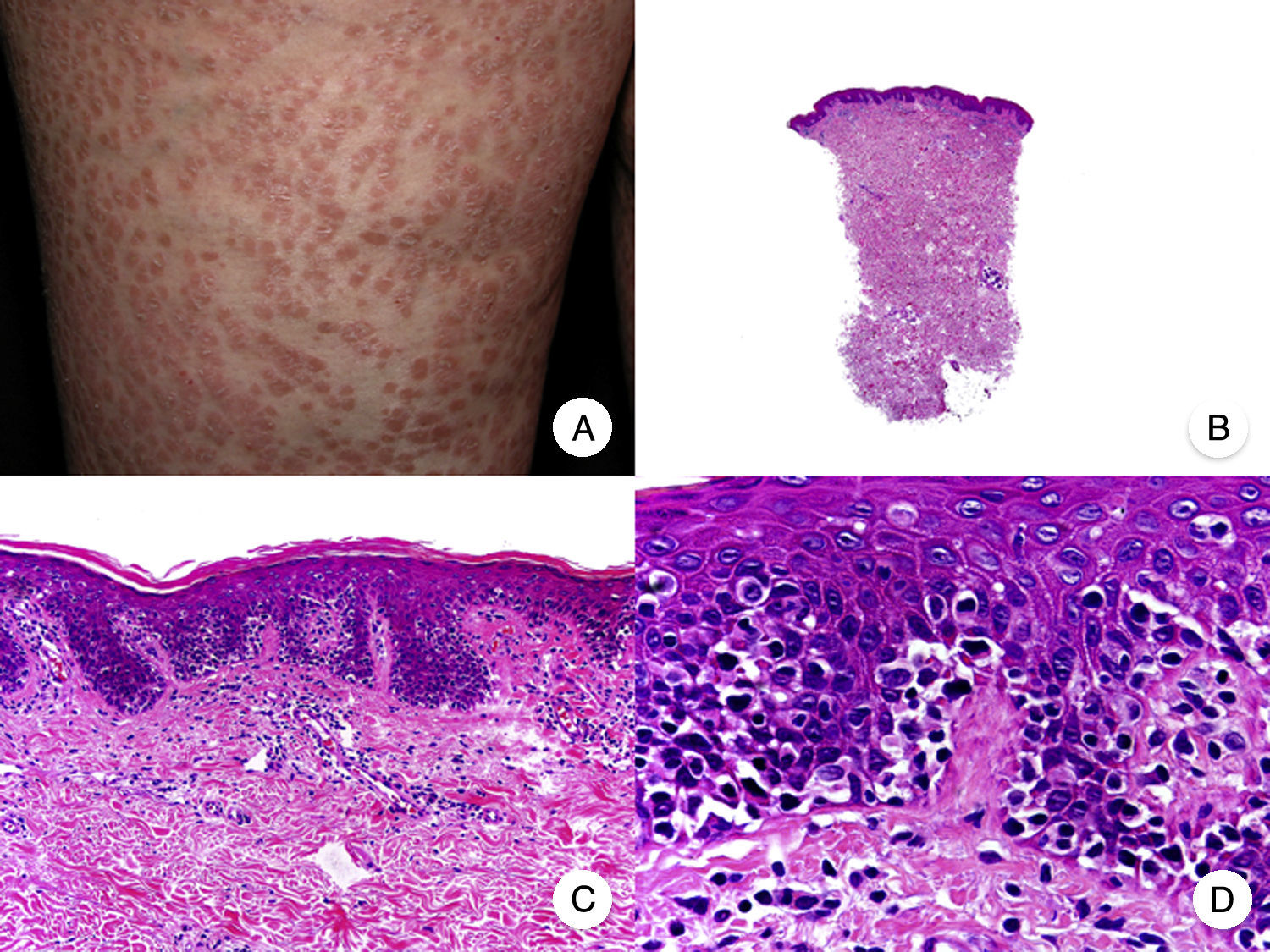
Micosis fungoide poiquilodérmicaLa MF poiquilodérmica, también denominada clásicamente poiquilodermia atrófica vascular, constituye una de las variantes más frecuentes de MF. Se caracteriza por la presencia de placas con atrofia, hiperpigmentación, hipopigmentación y telangiectasias que afectan a una amplia superficie cutánea, y parece tener un pronóstico favorable en comparación con otras variantes de la enfermedad, con buena respuesta a la fototerapia (fig. 9A)52. Clínicamente, las mamas en las mujeres y la región glútea en ambos sexos son las regiones cutáneas afectadas más frecuentemente por la MF poiquilodérmica.
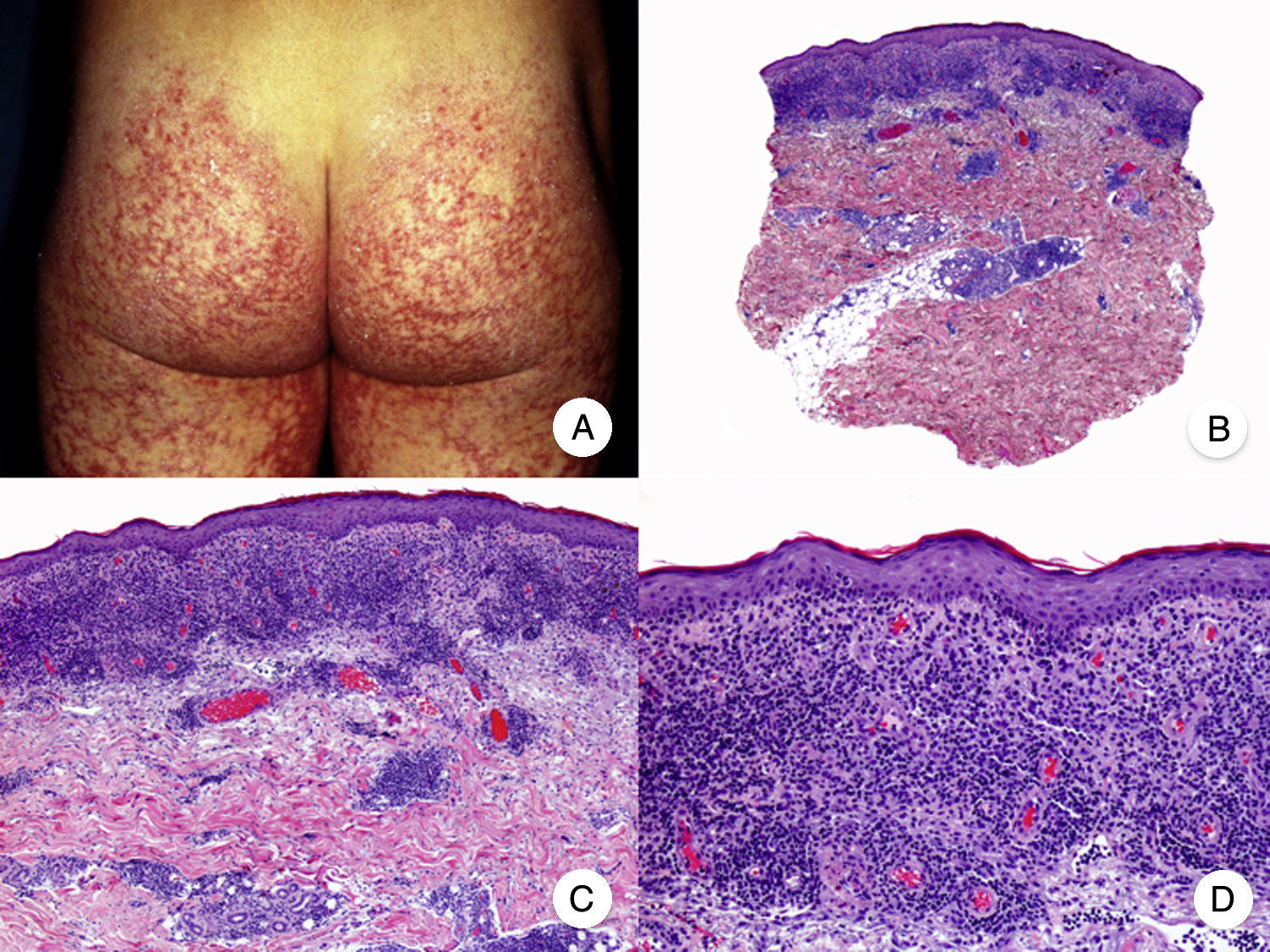
Micosis fungoide poiquilodérmica. A. Imagen clínica que muestra placas con atrofia, hiperpigmentación, hipopigmentación y telangiectasias afectando a ambas nalgas. B. Vista panorámica que muestra un infiltrado de tipo liquenoide en dermis papilar. C, D. A mayor aumento se observa atrofia epidérmica con aplanamiento de la unión dermoepidérmica, infiltrado de linfocitos atípicos con epidermotropismo y dilataciones, y dilataciones vasculares capilares en la dermis superficial.
Histopatológicamente se observa atrofia epidérmica y con aplanamiento de la unión dermoepidérmica, degeneración vacuolar de la hilera basal y un infiltrado de tipo liquenoide, compuesto por linfocitos atípicos con epidermotropismo. Otros hallazgos relevantes incluyen queratinocitos apoptóticos, incontinencia pigmentaria y dilataciones vasculares capilares en la dermis superficial (fig. 9 B-D). Sin embargo, los microabscesos de Darier-Pautrier no suelen estar presentes. Las células neoplásicas expresan a menudo un inmunofenotipo CD8+52.
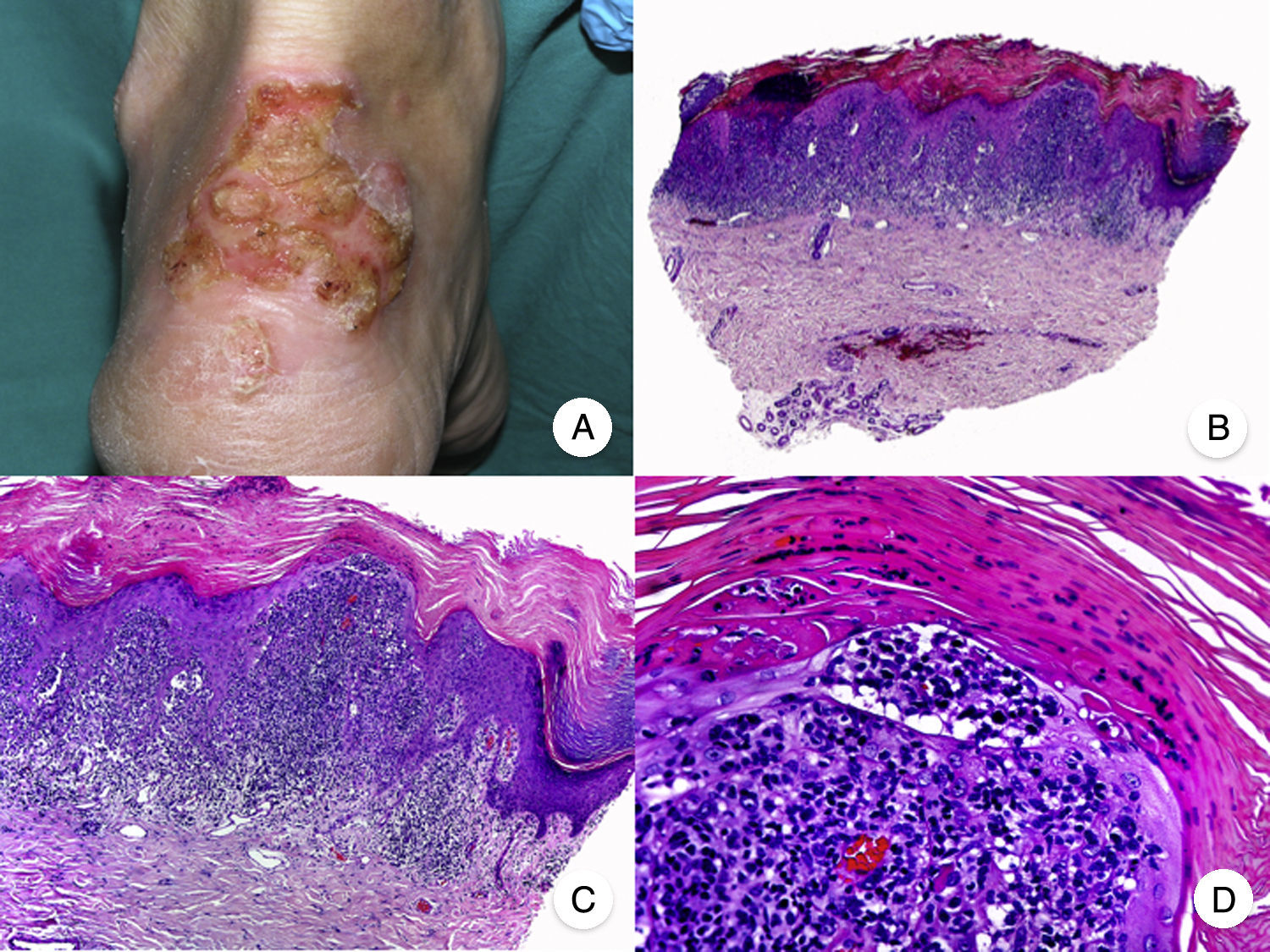
Micosis fungoide ampollarEl término MF bullosa o ampollar hace referencia a una variante clínico-patológica de MF definida por la presencia de lesiones vesiculoampollosas, originalmente descrita por Kaposi en 188753. Las lesiones vesiculoampollosas pueden ser flácidas o tensas, y afectan generalmente a una amplia superficie corporal en el tronco y las extremidades (fig. 10A). En ocasiones, la rotura de las ampollas provoca erosiones superficiales. Estas lesiones frecuentemente coexisten con las típicas de MF clásica, pudiendo constituir la primera manifestación de la enfermedad o aparecer en el transcurso de esta53,54. La presencia de lesiones ampollosas confinadas a palmas y plantas ha sido denominada MF dishidrótica55.
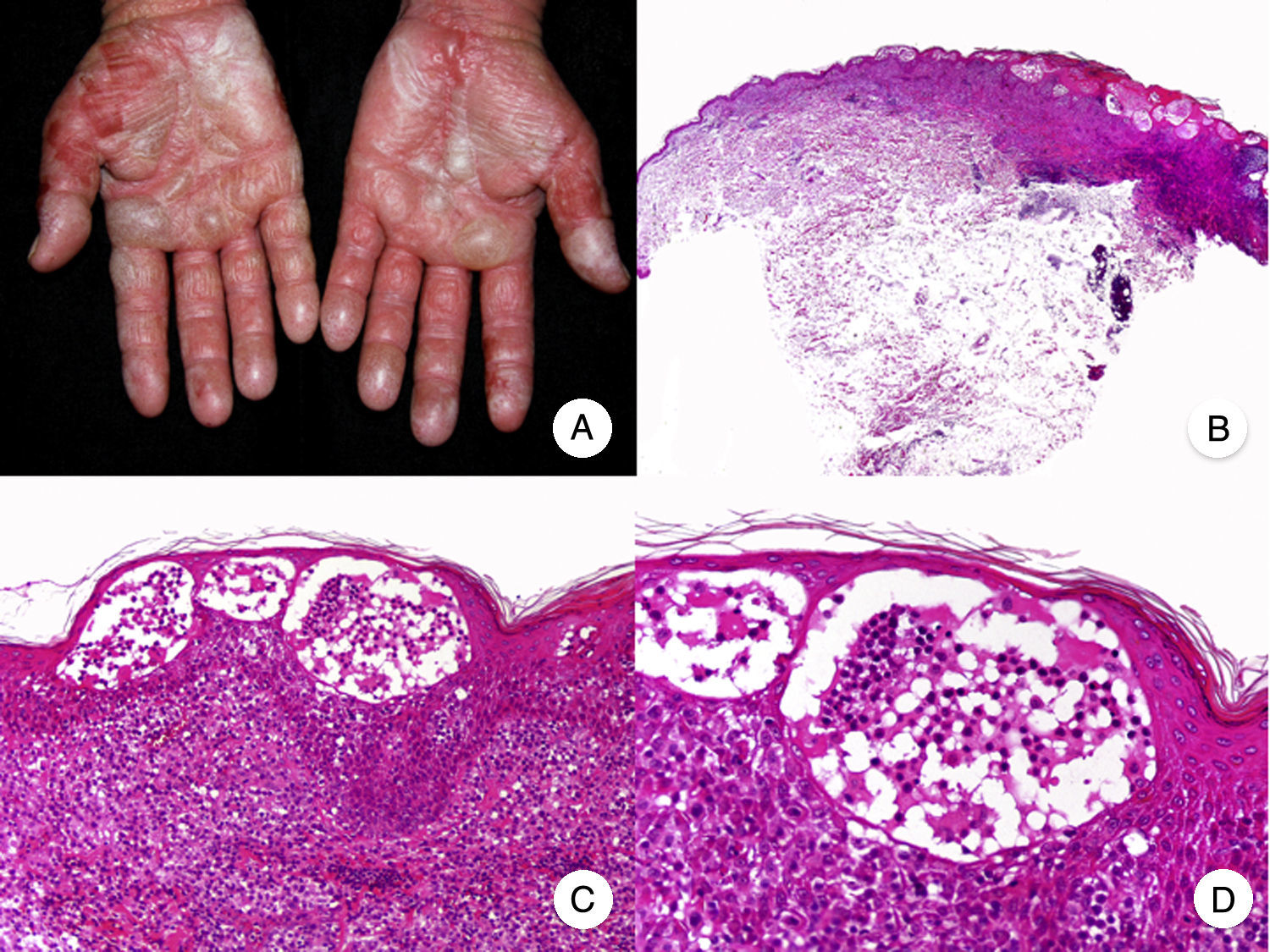
Desde el punto de vista histopatológico, la MF ampollar se caracteriza por la presencia de ampollas intraepidérmicas o subepidérmicas junto con los hallazgos histopatológicos propios de MF, incluyendo linfocitos atípicos, epidermotropismo y microabscesos de Darier-Pautrier (fig. 10 B-D). La inmunofluorescencia directa e indirecta es negativa, facilitando el diagnóstico diferencial con las enfermedades ampollosas autoinmunes. Otras causas de lesiones ampollosas, tales como fármacos e infecciones bacterianas o virales, deben tenerse también en cuenta en el diagnóstico diferencial53,56.
Se trata de una variante de mal pronóstico, con una supervivencia al año tras la aparición de las lesiones vesiculoampollosas de en torno al 50%53,57.
Micosis fungoide anetodérmicaLa anetodermia consiste en la pérdida progresiva de las fibras elásticas de la dermis, dando lugar clínicamente a la aparición de placas atróficas con una característica superficie apergaminada (fig. 11A). El desarrollo de anetodermia secundaria sobre lesiones clásicas de MF es excepcional, y hasta la fecha solo ha sido descrito en 2 pacientes58.
A. Imagen clínica que muestra un paciente con placas y nódulos atróficos de superficie apergaminada. B. Vista panorámica de un infiltrado difuso en toda la dermis. C. Detalle del infiltrado a mayor aumento, que muestra linfocitos atípicos y elastofagocitosis. D. Tinción de orceína que demuestra la ausencia de fibras elásticas en la dermis.
Desde el punto de vista histopatológico, las lesiones anetodérmicas observadas en estos pacientes mostraron hallazgos similares a los de la MF clásica, evidenciándose además la ausencia de fibras elásticas en la dermis mediante tinciones específicas como orceína o Van Gieson (fig. 11 B-D). A diferencia del síndrome de la piel laxa granulomatosa, la elastofagocitosis constituye un hallazgo focal y poco frecuente en la MF anetodérmica58.
Micosis fungoide hiperpigmentadaLa MF hiperpigmentada es una variante muy poco frecuente de MF que clínicamente se caracteriza por la presencia de máculas y placas hiperpigmentadas, que a menudo constituyen la única manifestación de la enfermedad. En ocasiones se asocia a otras variantes poco frecuentes de MF, pero los cambios pigmentarios observados no son atribuibles a alteraciones poiquilodérmicas o hiperpigmentación residual de lesiones preexistentes59.
Desde el punto de vista histopatológico se observa incontinencia pigmentaria, con abundantes gránulos de melanina en queratinocitos y células de Langerhans60, acompañados de numerosos melanófagos en la dermis papilar, así como de hallazgos típicos de MF clásica. Las células neoplásicas suelen localizarse en torno a la unión dermoepidérmica y muestran un inmunofenotipo característico CD8+61.
Esta variante parece tener un curso clínico indolente y poco agresivo59,61.
Micosis fungoide purpúricaEsta variante de MF se define clínicamente por la presencia de lesiones purpúricas persistentes, observándose histológicamente un infiltrado en banda compuesto fundamentalmente por linfocitos atípicos junto con hemosiderófagos y eritrocitos extravasados, que evoluciona a lesiones típicas de MF clásica, siendo más frecuente en varones62,63.
El diagnóstico diferencial con otras dermatosis purpúricas benignas constituye el reto fundamental debido al solapamiento clínico y en ocasiones también histopatológico, habiéndose planteado una posible relación entre la púrpura pigmentaria crónica y la MF63–65.
Micosis fungoide pustulosaEl término MF pustulosa hace referencia a una variante clínico-patológica extremadamente rara de MF que fue descrita inicialmente por Ackerman como una erupción vesiculopustulosa de largo tiempo de evolución que progresa clínicamente a una MF en placas clásica66. La afectación puede ser generalizada o estar limitada a las superficies palmoplantares67,68.
Histopatológicamente, esta variante muestra los hallazgos característicos de la MF clásica, como un infiltrado en banda compuesto por linfocitos atípicos, epidermotropismo y microabscesos de Darier-Pautrier, que se asocian a pústulas subcórneas con linfocitos atípicos, neutrófilos y eosinófilos en su interior68. La proporción entre las células neoplásicas y las inflamatorias es variable, predominando con frecuencia estas últimas66.
Micosis fungoide verrucosaLa MF verrucosa o hiperqueratósica constituye una variante muy poco frecuente de MF, caracterizada por la presencia de lesiones verrucosas e hiperqueratósicas localizadas o diseminadas, que pueden afectar a superficies acras y acompañarse de lesiones de MF clásica24,69,70. Es necesario realizar el diagnóstico diferencial con la variante palmar y plantar, confinada a dichos territorios. Desde el punto de vista histopatológico se observan los hallazgos característicos de la MF clásica, acompañados de un infiltrado inflamatorio perivascular en la dermis papilar, espongiosis y exocitosis en la epidermis, así como papilomatosis e hiperqueratosis71.
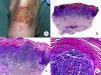
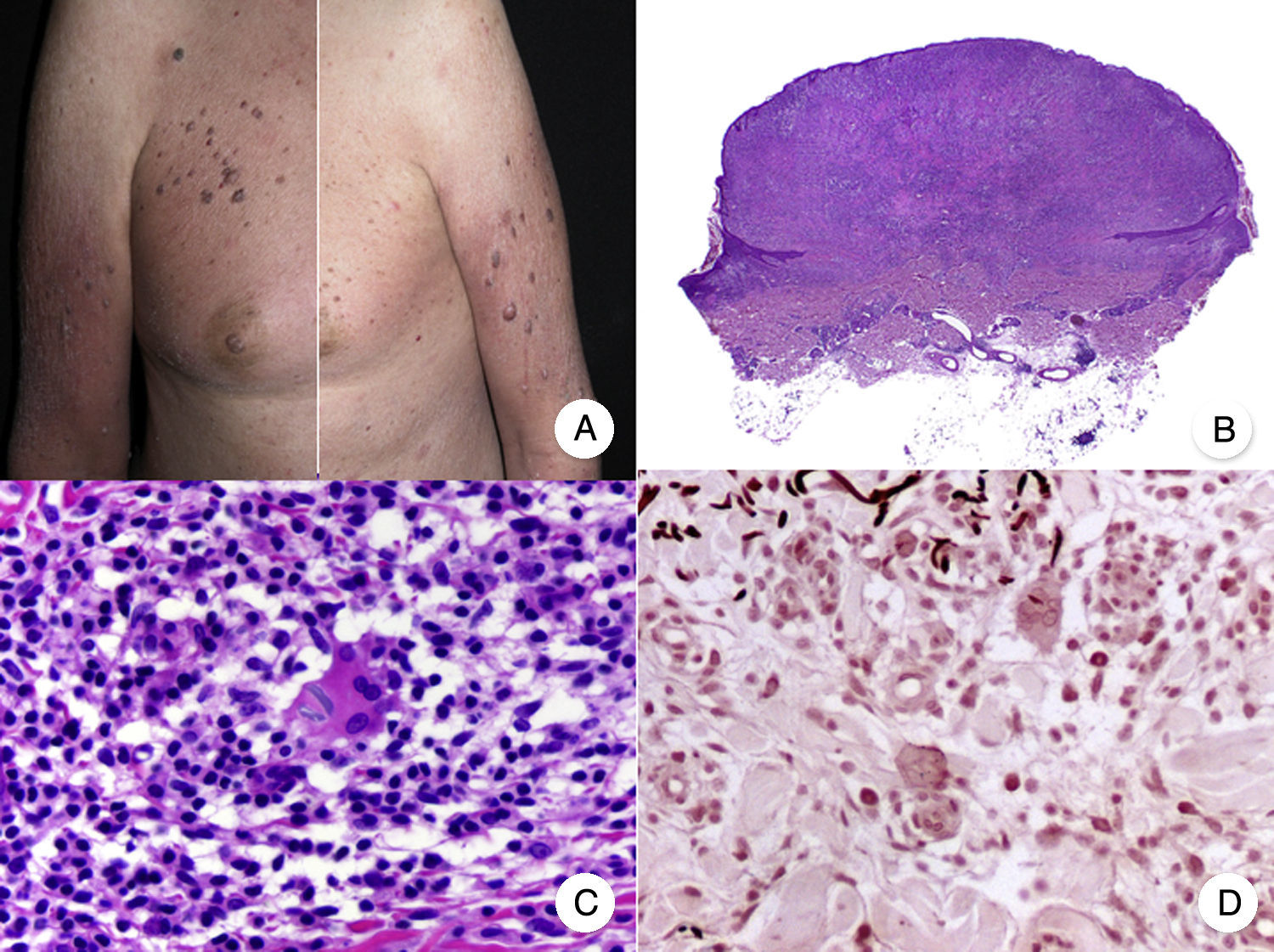
Variantes histopatológicas de micosis fungoideMicosis fungoide granulomatosaLa MF granulomatosa es una variante histopatológica de MF cuyo diagnóstico debe realizarse mediante biopsia cutánea, donde se observan granulomas de tipo sarcoideo perivasculares afectando a todo el espesor de la dermis, con un infiltrado acompañante compuesto por linfocitos atípicos, histiocitos y células gigantes multinucleadas72. El epidermotropismo solo está presente en la mitad de los casos, dificultando el diagnóstico de MF en ausencia de clínica característica45. En ocasiones se puede observar cierto grado de afectación folicular (fig. 12 B y C).
Micosis fungoide granulomatosa. A. Imagen clínica de lesiones de micosis fungoide en fase de parche y placa dispersas por el tronco y las extremidades. B. Vista panorámica que muestra granulomas perivasculares de tipo sarcoideo afectando a todo el espesor de la dermis. C. Detalle de un granuloma compuesto por linfocitos atípicos, histiocitos y células gigantes multinucleadas.
Clínicamente, la MF granulomatosa se caracteriza por la presencia de parches, placas y tumores similares a los de la MF clásica, pudiendo asociar alopecia cuando afecta al cuero cabelludo (fig. 12 A)73. Esta variante parece tener un peor pronóstico con respecto a la MF clásica, con pobre respuesta al tratamiento tópico, frecuente progresión y riesgo elevado de desarrollar un segundo linfoma73–75.
Aunque el diagnóstico diferencial con el síndrome de la piel laxa granulomatosa es eminentemente clínico, en la MF granulomatosa se observa una menor proporción de células gigantes multinucleadas, así como ausencia de elastolisis y elastofagocitosis24,49.
Micosis fungoide intersticialLa MF intersticial es una variante poco frecuente de MF, sin unas características clínicas específicas más allá de las propias de la MF clásica. Esta variante se define por los hallazgos histopatológicos, observándose un infiltrado de predominio linfocitario e histiocitos dispersos entre los haces de colágeno de la dermis, simulando la histología de un granuloma anular intersticial o la fase inflamatoria de la morfea (fig. 13)76–78. Otros hallazgos incluyen la presencia de epidermotropismo y el depósito de mucina en la dermis, siendo infrecuente observar microabscesos de Darier-Pautrier. El diagnóstico diferencial con el granuloma anular se basa en el reconocimiento de una población linfocitaria de tipo monoclonal, frente al infiltrado de predominio histiocitario observado en el granuloma anular, así como en la presencia de hallazgos característicos de MF clásica en otras lesiones, aunque ha sido descrita la coexistencia de ambos procesos en un mismo paciente79.
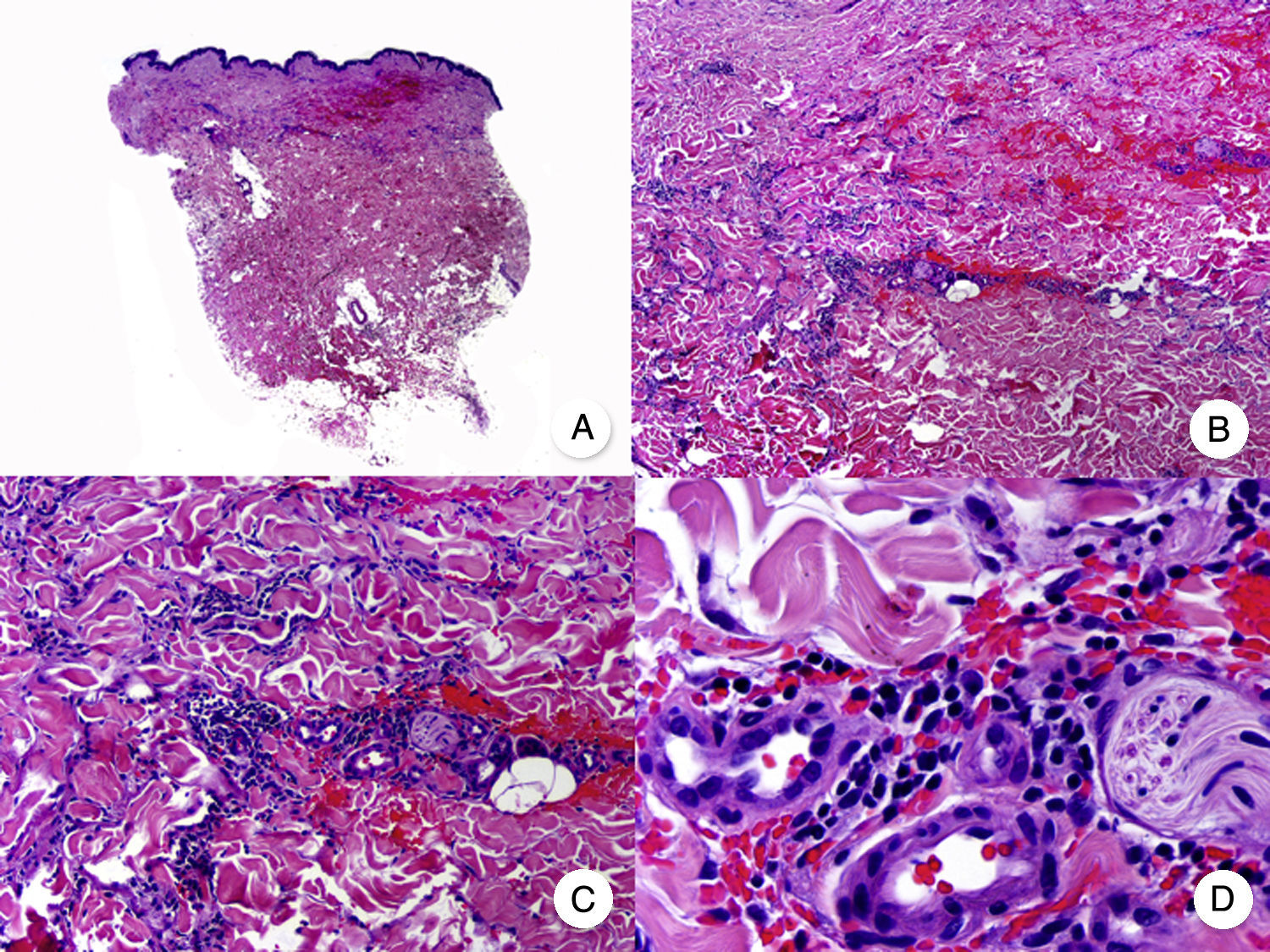
Micosis fungoide intersticial. A. Vista panorámica que muestra un leve infiltrado en la dermis. B. A mayor aumento se observa que el infiltrado se dispone siguiendo un patrón intersticial. C, D. Detalle del infiltrado de predominio linfocitario que se dispone entre los haces de colágeno y otras estructuras anexiales de la dermis.
La transformación en un linfoma de células grandes constituye una variante histopatológica de MF definida por la presencia de células grandes, pleomórficas, anaplásicas e inmunoblastos en un porcentaje mayor del 25% en el infiltrado neoplásico, siendo más frecuente en pacientes en estadio tumoral de la enfermedad80. La expresión de CD30 por las células grandes es variable, habiéndose relacionado su negatividad con un peor pronóstico (fig. 14)81.
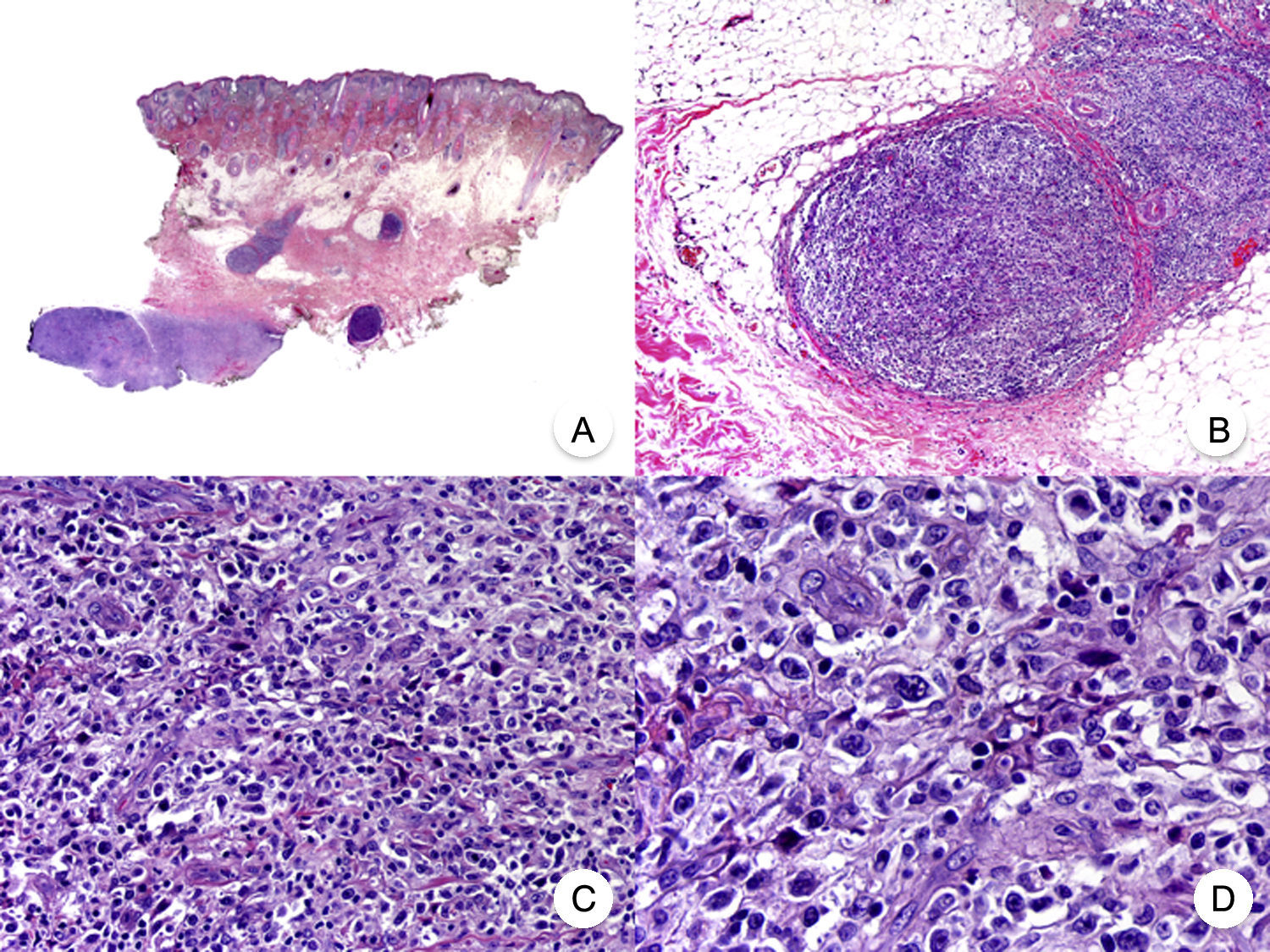
Micosis fungoide con transformación en células grandes. A. Vista panorámica que muestra una biopsia de cuero cabelludo con infiltración parcheada de la dermis. B. Detalle del infiltrado neoplásico en la hipodermis. C, D. A mayor aumento se observa que el infiltrado está compuesto por células neoplásicas grandes, pleomórficas y anaplásicas.
Esta variante histológica de MF debe distinguirse de otros linfomas primarios cutáneos con células grandes CD30+, como el linfoma anaplásico de células grandes y la papulosis linfomatoide. En la MF, el porcentaje de células grandes raramente alcanza el 75% necesario para realizar el diagnóstico de linfoma anaplásico de células grandes, y la expresión de la translocación IRF4, característica de este último, suele estar ausente82. El diagnóstico diferencial con la papulosis linfomatoide debe realizarse con base en los hallazgos clínicos, tales como la ausencia de regresión espontánea en las lesiones de MF83.
La transformación en un linfoma de células grandes confiere un peor pronóstico a la MF, asociándose a estadios terminales de la enfermedad81,82.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


