





Una mujer de 48 años, sin antecedentes médicos de interés, fue evaluada en el servicio de dermatología por la aparición de una lesión en el pubis de varios meses de evolución, que no había respondido al tratamiento con corticoides intralesionales. Negaba la existencia de una cicatriz o de un episodio de foliculitis previa.
Exploración físicaSe observaba una placa multinodular indurada, no dolorosa, de superficie lisa y límites netos de 4cm de diámetro localizada en el pubis (fig. 1). Además, se observaron 2 lesiones papuloeritematosas menores de 1cm localizadas bilateralmente en el escote. No presentaba síntomas sistémicos y no se palparon adenopatías.
Histopatología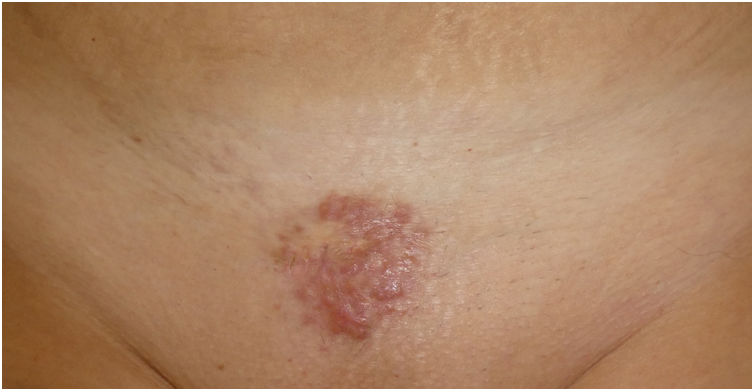
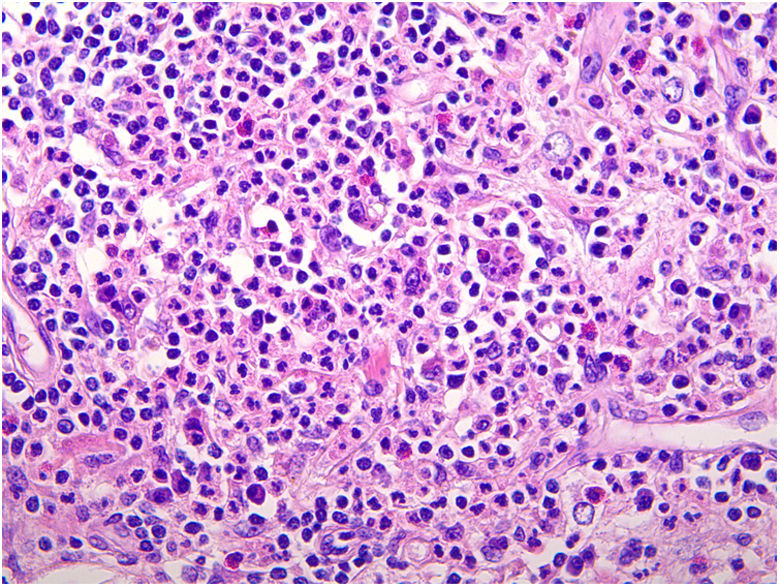
La biopsia mostró una epidermis sin alteraciones y un denso infiltrado polimorfo en la dermis media y profunda, así como un aumento de la trama de colágeno y de la vascularización (fig. 2). Se observaba el fenómeno de emperipolesis.
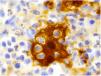
La inmunohistoquímica demostró una positividad para CD68, S100 (fig. 3) y ciclina D1 y resultó negativa para CD1a.
Otras pruebas complementarias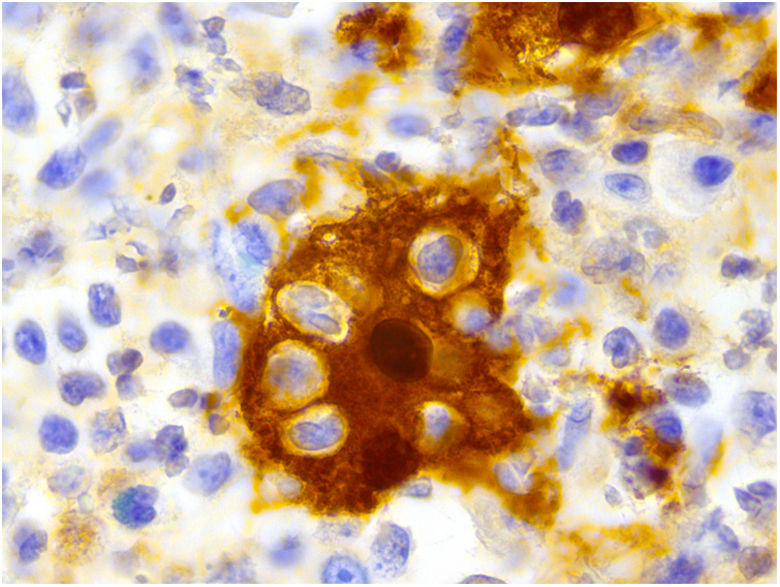
Se realizó un estudio de extensión con una TAC y una analítica completa, que no mostraron alteraciones. Además, se solicitaron serologías con resultado negativo para VEB, CMV, VH6, VH8, VIH y lúes y cultivos en piel negativos para micobacterias y treponemas.
¿Cuál es su diagnóstico?
DiagnósticoEnfermedad cutánea de Rosai Dorfman.
Evolución y tratamientoLas lesiones fueron extirpadas quirúrgicamente. La paciente presentó una nueva lesión en escote a los 4 meses de seguimiento que volvió a ser extirpada, con los mismos hallazgos histológicos.
ComentarioLa enfermedad de Rosai Dorfman es un transtorno proliferativo histiocítico de células no de Langerhans que clásicamente cursa con la presencia de adenopatías bilaterales masivas y no dolorosas, así como con una clínica sistémica, una elevación de los reactantes de fase aguda e hipergammaglobulinemia. Hasta un 40% de los pacientes pueden mostrar una clínica extraganglionar, donde en la piel se afecta en torno al 10%1.
Existe una forma únicamente cutánea, que tiene una edad media de aparición mayor a la forma clásica (43,5 años en la forma cutánea frente a 20,6 años en la forma clásica) y un predominio en la mujer1.
La presentación clínica más frecuente es la pápulo-nodular, si bien se han descrito lesiones muy variadas (placas induradas, tumores, lesiones acneiformes, xantogranulomatosas y ulceraciones). Esto hace que el diagnóstico diferencial sea muy variado, incluyendo otras proliferaciones histiocitarias, inflamatorias y tumorales (linfoma cutáneo, sarcoma de Kaposi, dermatofibrosarcoma protuberans…), así como infecciones cutáneas por micobacterias. La localización predominantemente es en el tórax y, con frecuencia, hay más de una lesión en el momento del diagnóstico1.
Se ha relacionado con infecciones virales, en especial con la familia herpes virus y mutaciones en MAP2K1/ERK, proteínas que forman parte de una de las vías reguladoras de la proliferación y la supervivencia celular. Se ha establecido su asociación con enfermedades autoinmunes, tales como el lupus eritematoso sistémico y la artritis idiopática juvenil, así como con neoplasias de estirpe linfoide y mieloide. Hay descrita una forma familiar con mutaciones en SLC29A3 en la línea germinal. Este gen se ha implicado en el síndrome H y en la hipertricosis pigmentada con diabetes mellitus insulinodependiente. Por último, se han detectado niveles elevados de IgG4, sobre todo en la forma clásica con una afectación visceral, que abre el debate del posible encuadre de esta entidad dentro de la enfermedad relacionada con IgG42.
Su diagnóstico se realiza con el estudio histopatológico de la lesión, así como un conjunto de pruebas complementarias que descarten una enfermedad a otro nivel. Estas incluyen pruebas de imagen, generalmente TAC y estudios analíticos con hemograma, bioquímica, función renal y hepática, ANA, complemento, proteinograma y serologías para VEB, CMV, VH6, VH8, VIH y lúes3.
La biopsia muestra un infiltrado dérmico denso de histiocitos con linfocitos, células plasmáticas y neutrófilos dispersos. Un dato clave en el diagnóstico es la presencia de fenómeno de emperipolesis. La inmunohistoquímica refleja el origen de la estirpe celular que conforma la lesión, siendo positiva para CD14, S100, CD68 y CD163 (marcadores de histiocitos) y negativa para CD1a y langerhina (marcadores de células de Langerhans) y factor XIIIa1.
La exéresis quirúrgica es una alternativa terapéutica4, así como el uso de corticoides orales e intralesionales, metotrexato5, imiquimod tópico6 y crioterapia, de los que se han descrito casos aislados. El pronóstico en general es bueno, pudiendo haber remisión espontánea de las lesiones hasta en un 20% de las ocasiones4.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


