






El tumor de células granulares cutáneo (TCG) o de Abrikossoff es una neoplasia benigna e infrecuente, probablemente de origen neural a partir de las células de Schwann. Clínicamente se manifiesta como un nódulo solitario y asintomático, y su etiopatogenia ha estado ampliamente debatida en la literatura.
ObjetivosAnalizar las características clínicas, histológicas e inmunohistoquímicas asociadas a este tumor y determinar si estos hallazgos se corresponden con lo descrito hasta el momento en la literatura.
MétodosSe realiza un estudio retrospectivo de treinta y cuatro casos con diagnóstico histológico de TCG donde se analizan las características clínicas (localización, edad, sexo, tiempo de evolución y sospecha diagnóstica), histológicas (delimitación, atipia, mitosis, afectación de anejos cutáneos, hiperplasia seudoepiteliomatosa y presencia de los recientemente descritos cuerpos pústulo-ovoides [CPO]) e inmunohistoquímicas (tinción S-100 en 16 casos seleccionados aleatoriamente).
ResultadosEl 58,82 % fueron hombres y el 41,18 % mujeres. La edad media fue de 31,74 años. La localización más frecuente fue la cavidad oral (61,76 %). El diagnóstico clínico más comúnmente sospechado fue el de fibroma (17,65 %). En un 85,29 % se presentaba como una lesión mal delimitada y difusa. La hiperplasia seudoepiteliomatosa estuvo presente en el 58,82 %. Se encontraron atipias nucleares en el 29,41 % y mitosis en un 20,59 %. Hubo un caso maligno (2,94 %) y dos se consideraron atípicos (5,88 %). Los CPO estuvieron presentes en el 47,06 %. La tinción con S-100 fue positiva en el 100 % de los casos analizados.
ConclusionesNuestra serie confirmaría las características descritas hasta el momento para el TCG salvo con algunas peculiaridades y apoyaría la existencia de los CPO como un nuevo hallazgo histológico adicional para el diagnóstico de este tumor.
Granular cell tumor (GCT), also known as Abrikossoff tumor, is an uncommon benign neoplasm, probably of neural origin derived from Schwann cells. It presents clinically as a solitary asymptomatic nodule and its pathogenesis has been the subject of much debate in the literature.
ObjectivesWe aimed to analyze the clinical, histologic, and immunohistochemical characteristics associated with this tumor and to determine whether these findings correspond to those reported to date in the literature.
MethodsIn this retrospective study of 34 patients with histologic diagnosis of GCT, we analyzed clinical characteristics (site, age, sex, duration, and suspected diagnosis), histological findings (border, cell atypia and mitoses, involvement of adnexal structures, pseudoepitheliomatous hyperplasia, and presence of the recently described pustulo-ovoid bodies), and immunohistochemical findings (S-100 staining in 16 randomly selected cases).
ResultsIn total, 58.82 % were men and 41.18 % were women, and the mean age was 31.74 years. The most common site was the oral cavity (61.76 %). The most frequently suspected clinical diagnosis was fibroma (17.65 %). The lesion was poorly defined and diffuse in 85.29 %. Pseudoepitheliomatous hyperplasia was present in 58.82 %. Nuclear atypia was found in 29.41 % and mitoses in 20.59 %. One case was considered malignant (2.94 %) and 2 atypical (5.88 %). Pustulo-ovoid bodies were present in 47.06 % of the cases and S-100 staining was positive in all cases analyzed.
ConclusionsOur series confirms the characteristics described previously for GCT, except for certain peculiarities, and supports the presence of pustulo-ovoid bodies as an additional histologic finding for diagnosis of this tumor.
El tumor de células granulares (TCG), también conocido como tumor de Abrikossoff, es una neoplasia benigna e infrecuente, probablemente de origen neural derivada de las células de Schwann1. El caso típico se presenta como una lesión nodular, de crecimiento lento, solitaria e indolora, localizada a nivel cutáneo y subcutáneo (43 %), en la lengua (23 %) o en cualquier otra localización anatómica2 (fig. 1). Puede afectar a todas las edades, razas o sexos, pero es más frecuente entre la tercera y quinta décadas de la vida, en mujeres y en la población de raza negra2.
. A. Nódulo polipoide con aspecto de fibroma en el dorso de la lengua. B. Nódulo violáceo bien delimitado en el dorso lingual. C. Nódulo del color de la lengua mal delimitado.")
El diagnóstico se establece mediante biopsia y, en general, no se sospecha clínicamente por ser ésta poco específica. Los casos malignos representan un 1-2 % y generalmente tienen un desenlace fatal3. Estos casos no se han descrito en niños.
Los pacientes con lesiones múltiples se dan en el 5-25 % de los casos y también son infrecuentes en niños4-6. Existen casos familiares de lesiones múltiples que plantean la posibilidad de que algunos pacientes tengan una susceptibilidad hereditaria. También se han descrito casos congénitos7 y la asociación en niños de tumores cutáneos múltiples y anomalías sistémicas musculoesqueléticas, cardiovasculares y neurológicas8,9.
El TCG, que fue inicialmente descrito por Abrikossoff en 19261, tiene una apariencia histológica característica, fácilmente reconocible con el microscopio óptico. Sin embargo, la patogenia de este tumor ha sido tema de investigación y debate durante muchos años sin haberse llegado hoy en día a aclarar su origen. El TCG fue inicialmente denominado por Abrikossoff como «mioblastoma de células granulares» porque pensó que tenía una apariencia histológica similar a las células del músculo estriado1. Estudios posteriores apoyaron una diferenciación neural, hipótesis propuesta por primera vez por Feyrter en 19351. En 1962, usando el microscopio electrónico, Fisher y Wechsler postularon una diferenciación a partir de las células de Schwann, que es actualmente la hipótesis aceptada para el TCG1.
Al microscopio óptico las células que constituyen el TCG son grandes, poligonales o redondas, de núcleo vesiculoso central, citoplasma eosinófilo y granular, y en el epitelio suprayacente se asocia con una hiperplasia seudoepiteliomatosa variable. Las células tumorales individuales están frecuentemente separadas por finas bandas de tejido conectivo (fig. 2). Los gránulos prominentes que se observan en el interior de las células representan lisosomas o un componente del aparato de Golgi. Los estudios histoquímicos de rutina demuestran que los gránulos son positivos para el ácido periódico de Schiff (PAS) y el azul luxol, y resistentes a la diastasa, lo que indica la presencia de mielina en el interior del tumor. La mayoría de los TCG son S-100 y CD57 positivos, además de mostrar una fuerte positividad también para una enolasa neuronal específica (NSE) y vimentina. Se ha demostrado la presencia de glucoproteína asociada a la mielina. Recientemente se ha descrito la presencia de «cuerpos pústulo-ovoides (CPO) de Milian» como un hallazgo histológico constante. Estos CPO se observan al microscopio óptico como gránulos eosinófilos de gran tamaño (4-5μm), redondeados y rodeados por un halo claro (fig. 3). Su significación y frecuencia en este tumor no están bien aclarada10.
 ocupando la dermis (hematoxilina-eosina original, ×10). B. TCG compuesto por células de gran tamaño, poligonales y fusocelulares con citoplasma eosinófilo y granular (hematoxilina-eosina, ×100).")
.")
Cabe señalar que las células granulares pueden estar presentes también en diversos tumores como el carcinoma basocelular, los fibroxantomas, los fibroxantomas atípicos11,12, el dermatofibroma13, los dermatomiofibromas, el dermatofibrosarcoma protuberans, los nevos melanocíticos14, los rabdomiomas, los angiosarcomas cutáneos y los leiomiosarcomas cutáneos. Este hallazgo es frecuente e inespecífico y afecta a una parte de la lesión, permitiendo, mediante criterios morfológicos e inmunohistoquímicos, una correcta diferenciación del verdadero TCG. En ocasiones, cambios reactivos asociados con un trauma quirúrgico pueden simular un TCG6.
Recientemente se han descrito casos de TCG con afectación de la unión dermoepidérmica que imitarían una neoplasia melanocítica14. Esto podría complicar aún más el diagnóstico diferencial con el melanoma. Ante esta situación desempeñarían un papel muy importante las tinciones de inmunohistoquimia (que en el TCG serían negativas para HMB-45 y Melan-A, y positivas para calretinina e inhibina).
Es importante mencionar que existe una variedad de TCG no neural también llamada «TCG polipoide primitivo» que no muestra una clara línea de diferenciación. Parece ser que esta categoría posee unas características histológicas atípicas que permitirían encuadrarlo en una nueva entidad y distinguirlo del TCG de origen neural15,16.
ObjetivosSe pretenden analizar las características clínicas, epidemiológicas, histológicas e inmunohistoquímicas asociadas a este tumor y determinar si estos hallazgos se corresponden con lo descrito hasta el momento en la literatura.
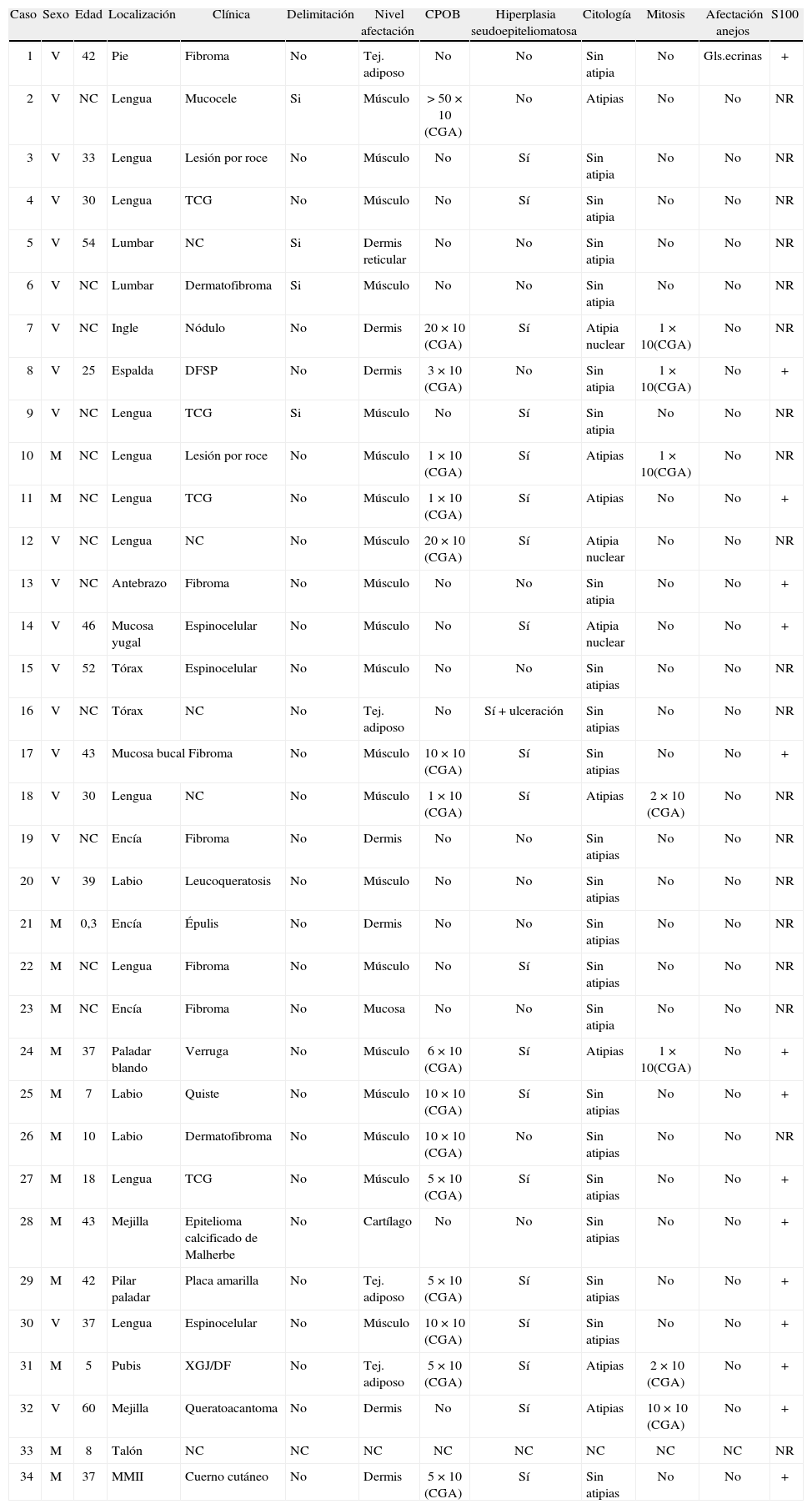
Material y métodosSe revisan de forma retrospectiva 34 casos con diagnóstico anatomopatológico de TCG de piel y mucosas recogidos en el servicio de Dermatología del Consorcio Hospital General Universitario de Valencia entre los años 1980 y 2007. Se analizan las características clínicas en cuanto a localización del tumor, la edad de aparición, el sexo, el tiempo de evolución hasta el diagnóstico y el diagnóstico clínico sospechado. También se analizan las características histológicas en cuanto a la arquitectura, la atipia celular y nuclear, el número de mitosis, el grado de afectación de anejos cutáneos, la presencia de hiperplasia seudoepiteliomatosa y la presencia de CPO. Se realizan estudios de inmunohistoquimia con la tinción S-100 en 16 casos seleccionados de forma aleatoria.
Todos los casos fueron fijados en formol al 10 % tamponado (WWR International), embebidos en parafina y procesados de forma estándar. Las secciones se prepararon con hematoxilina-eosina y en todos los casos se realizaron tinciones con PAS que resaltaron los gránulos intracitoplasmáticos.
ResultadosHallazgos clínicosDel total de 34 pacientes, 20 (58,82 %) fueron hombres y 14 (41,18 %) mujeres, todos ellos de raza caucásica.
La edad de los pacientes osciló entre los 3 meses y los 60 años, con una mediana de 37 años y una edad media de 31,74 años, no pudiéndose determinar en 12 de los casos (tabla 1). El TCG se presentó mayoritariamente como un nódulo solitario, generalmente asintomático que afectaba sobre todo a la cavidad oral en 21 casos (61,76 %) (figs. 1 y 2), a las extremidades en 5 (17,65 %), a la mejilla en 2 (5,88 %), a la espalda en 3 (8,82 %), al tronco en 2 (5,88 %) y al pubis en 1 (2,94 %) (tabla 1). El tamaño de la lesión, en los casos en que este dato pudo ser recogido, osciló entre pocos milímetros y varios centímetros, no pudiéndose determinar en la mayoría de los casos, así como tampoco el tiempo de evolución que variaba en algunos casos entre meses y años.
Características de los pacientes
| N.° de pacientes (%) | |
| Pacientes evaluados | 34 (100) |
| Tumores evaluados | 34 (100) |
|
|
|
|
|
|
En todos los casos la escisión quirúrgica fue el tratamiento de elección, no habiéndose detectado recidiva o metástasis en ninguno de los casos hasta la fecha (tiempo transcurrido desde el diagnóstico entre 4-27 años).
El diagnóstico clínico inicial de sospecha de TCG sólo se realizó en 5 casos (14,71 %), todos ellos localizados en la cavidad oral; 4 casos en la lengua y 1 caso en la encía. El diagnóstico clínico más comúnmente sospechado fue de fibroma en 6 de los 34 casos (17,65 %) (fig. 1), seguido por el de TCG en 5 (14,71 %) y el de carcinoma espinocelular en 4 (11,76 %). Menos frecuentemente se sospechó un dermatofibroma, 3 casos (8,82 %); una lesión por roce, 2 casos (5,88 %) y un xantogranuloma, 2 casos (5,88 %). En último lugar, y de forma más anecdótica, aparecen mucocele, nódulo, dermatofibrosarcoma protuberans, leucoqueratosis, verruga, quiste, epitelioma calcificado de Malherbe y queratoacantoma como sospecha diagnóstica inicial representando 1 de los 34 casos (2,94 %).
Hallazgos histológicosDesde el punto de vista arquitectural, el TCG aparece como una lesión mal delimitada y de distribución difusa en la dermis en 29 de los 34 casos (85,29 %). Cuando afecta a la boca y la cavidad oral alcanza el músculo estriado, y cuando afecta a la piel y al tejido subcutáneo llega hasta el panículo adiposo.
La hiperplasia seudoepiteliomatosa está presente en 20 de los 34 casos (58,82 %) de forma moderada, confundiéndose en ocasiones el diagnóstico histológico con un carcinoma espinocelular cuando es muy intensa y sólo se dispone de una biopsia superficial para su examen (fig. 4). Las células tumorales eran de gran tamaño, poligonales, redondas o fusocelulares y con un citoplasma amplio, eosinófilo, con abundantes y finos gránulos en su interior (fig. 2). El núcleo celular era generalmente de pequeño y mediano tamaño, situado centralmente y con una apariencia vesicular o hipercromática, con uno o múltiples nucléolos pequeños y eosinófilos en su interior. El pleomorfismo nuclear fue fundamentalmente focal y muy leve, aunque encontramos atipias nucleares en 10 de los 34 casos (29,41 %). Se encontraron mitosis en 7 de los 34 casos (20,59 %) con un rango de actividad mitótica entre 1 y 10 (media: 2,57, mediana: 1) que generalmente oscilaba entre 1 ó 2 mitosis por campo de 10 aumentos, salvo en un caso que se diagnosticó de TCG maligno (1/34) (2,94 %) por su gran extensión clínica e histológica, presencia de atipias nucleares y mitosis en número superior a 2 por campo de 10 aumentos, y pleomorfismo nuclear. Dos casos se consideraron como atípicos (5,88 %) por cumplir menos de estas tres características histológicas (casos 31 y 18). Sólo hubo afectación de los anejos en un caso; en concreto afectación de las glándulas ecrinas, representando un 2,94 %. No se detectó afectación linfovascular, necrosis, ni invasión perineural en ninguno de los casos.
 simulando un carcinoma espinocelular (hematoxilina-eosina, ×10). B. Imagen en detalle de la HSE con las células granulares en dermis papilar (hematoxilina-eosina, ×200).")
La presencia de «CPO de Milian» se observó en 16 de los 34 casos analizados (47,06 %). Un caso (6,25 %) tuvo más de 50 CPO por campo de 10 aumentos. Seis casos (37,50 %) tuvieron entre 10 y 30 CPO por campo de 10 aumentos y 9 casos (56,25 %) tuvieron menos de 10 CPO por campo de 10 aumentos (fig. 3; tablas 1 y 2).
Resumen de datos clínicos, histológicos e inmunohistoquímicos del TCG
| Caso | Sexo | Edad | Localización | Clínica | Delimitación | Nivel afectación | CPOB | Hiperplasia seudoepiteliomatosa | Citología | Mitosis | Afectación anejos | S100 |
| 1 | V | 42 | Pie | Fibroma | No | Tej. adiposo | No | No | Sin atipia | No | Gls.ecrinas | + |
| 2 | V | NC | Lengua | Mucocele | Si | Músculo | >50 × 10 (CGA) | No | Atipias | No | No | NR |
| 3 | V | 33 | Lengua | Lesión por roce | No | Músculo | No | Sí | Sin atipia | No | No | NR |
| 4 | V | 30 | Lengua | TCG | No | Músculo | No | Sí | Sin atipia | No | No | NR |
| 5 | V | 54 | Lumbar | NC | Si | Dermis reticular | No | No | Sin atipia | No | No | NR |
| 6 | V | NC | Lumbar | Dermatofibroma | Si | Músculo | No | No | Sin atipia | No | No | NR |
| 7 | V | NC | Ingle | Nódulo | No | Dermis | 20 × 10 (CGA) | Sí | Atipia nuclear | 1 × 10(CGA) | No | NR |
| 8 | V | 25 | Espalda | DFSP | No | Dermis | 3 × 10 (CGA) | No | Sin atipia | 1 × 10(CGA) | No | + |
| 9 | V | NC | Lengua | TCG | Si | Músculo | No | Sí | Sin atipia | No | No | NR |
| 10 | M | NC | Lengua | Lesión por roce | No | Músculo | 1 × 10 (CGA) | Sí | Atipias | 1 × 10(CGA) | No | NR |
| 11 | M | NC | Lengua | TCG | No | Músculo | 1 × 10 (CGA) | Sí | Atipias | No | No | + |
| 12 | V | NC | Lengua | NC | No | Músculo | 20 × 10 (CGA) | Sí | Atipia nuclear | No | No | NR |
| 13 | V | NC | Antebrazo | Fibroma | No | Músculo | No | No | Sin atipia | No | No | + |
| 14 | V | 46 | Mucosa yugal | Espinocelular | No | Músculo | No | Sí | Atipia nuclear | No | No | + |
| 15 | V | 52 | Tórax | Espinocelular | No | Músculo | No | No | Sin atipias | No | No | NR |
| 16 | V | NC | Tórax | NC | No | Tej. adiposo | No | Sí + ulceración | Sin atipias | No | No | NR |
| 17 | V | 43 | Mucosa bucal Fibroma | No | Músculo | 10 × 10 (CGA) | Sí | Sin atipias | No | No | + | |
| 18 | V | 30 | Lengua | NC | No | Músculo | 1 × 10 (CGA) | Sí | Atipias | 2 × 10 (CGA) | No | NR |
| 19 | V | NC | Encía | Fibroma | No | Dermis | No | No | Sin atipias | No | No | NR |
| 20 | V | 39 | Labio | Leucoqueratosis | No | Músculo | No | No | Sin atipias | No | No | NR |
| 21 | M | 0,3 | Encía | Épulis | No | Dermis | No | No | Sin atipias | No | No | NR |
| 22 | M | NC | Lengua | Fibroma | No | Músculo | No | Sí | Sin atipias | No | No | NR |
| 23 | M | NC | Encía | Fibroma | No | Mucosa | No | No | Sin atipia | No | No | NR |
| 24 | M | 37 | Paladar blando | Verruga | No | Músculo | 6 × 10 (CGA) | Sí | Atipias | 1 × 10(CGA) | No | + |
| 25 | M | 7 | Labio | Quiste | No | Músculo | 10 × 10 (CGA) | Sí | Sin atipias | No | No | + |
| 26 | M | 10 | Labio | Dermatofibroma | No | Músculo | 10 × 10 (CGA) | No | Sin atipias | No | No | NR |
| 27 | M | 18 | Lengua | TCG | No | Músculo | 5 × 10 (CGA) | Sí | Sin atipias | No | No | + |
| 28 | M | 43 | Mejilla | Epitelioma calcificado de Malherbe | No | Cartílago | No | No | Sin atipias | No | No | + |
| 29 | M | 42 | Pilar paladar | Placa amarilla | No | Tej. adiposo | 5 × 10 (CGA) | Sí | Sin atipias | No | No | + |
| 30 | V | 37 | Lengua | Espinocelular | No | Músculo | 10 × 10 (CGA) | Sí | Sin atipias | No | No | + |
| 31 | M | 5 | Pubis | XGJ/DF | No | Tej. adiposo | 5 × 10 (CGA) | Sí | Atipias | 2 × 10 (CGA) | No | + |
| 32 | V | 60 | Mejilla | Queratoacantoma | No | Dermis | No | Sí | Atipias | 10 × 10 (CGA) | No | + |
| 33 | M | 8 | Talón | NC | NC | NC | NC | NC | NC | NC | NC | NR |
| 34 | M | 37 | MMII | Cuerno cutáneo | No | Dermis | 5 × 10 (CGA) | Sí | Sin atipias | No | No | + |
CGA: campo de gran aumento; CPO: cuerpos pústulo-ovoides; M: mujer; MMII: miembros inferiores; NC: no conocido; NR: no realizado; TCG: tumor de células granulares; V: varón.
Se realizaron tinciones con S-100 en 16 de los 34 casos elegidos de forma aleatoria, encontrándose una fuerte positividad en todos ellos (100 %) (fig. 5; tablas 1 y 2).
Discusión.")
En nuestra serie el TCG aparece como un tumor que puede afectar a todas las edades y sexos, siendo más frecuente en el sexo masculino, y entre la segunda y la quinta décadas de la vida, a diferencia de lo que se viene describiendo en la literatura donde siempre se ha considerado un tumor más frecuente en mujeres, y entre la tercera y quinta décadas. No obstante, llama la atención un número no desdeñable de casos en niños y adolescentes (17,65 %). Tampoco hubo ningún caso de raza negra2. En nuestra serie fue más frecuente la localización en las mucosas de la cavidad oral que en la piel y el tejido subcutáneo.
En ningún caso se dio la presencia de tumores múltiples, casos familiares o lesiones congénitas. Asimismo, tampoco concurrieron otras anomalías sistémicas asociadas al TCG en los niños como se han descrito en la literatura8,9.
No se pudieron realizar tinciones con S-100 en todos los casos, pero aquellos en los que se realizaron mostraron una positividad marcada para S-100. Esto apoya el origen neural del TCG y descarta incluirlos en la variedad descrita por Le Boit et al, y posteriormente apoyada por Chaudhry y Calonje, de TCG dérmico no neural o clásicamente conocido como TCG primitivo polipoide que es principalmente S-100 negativo y muestra unas características histológicas que permiten diferenciarlo del TCG de origen neural. Dichas características serían: arquitectura polipoide, collarete epidérmico, localización en la dermis papilar, buena delimitación, hiperplasia epidérmica mínima o ausente, atipia citológica (de leve a moderada frente a focal a difusa) y aumento de la actividad mitótica. Pese a ello tienen un comportamiento biológico benigno y un buen pronóstico15,16.
Sólo uno de los casos se consideró maligno, no conociéndose la evolución del paciente. El diagnóstico de TCG maligno se estableció en función de los criterios propuestos por Enzinger y Weiss que incluyen entre otros: núcleo vesicular con nucléolo prominente, aumento de la actividad mitótica (más de 2 mitosis por 10 campos de gran aumento [CGA]), relación núcleo/citoplasma elevada y pleomorfismo3. El término atípico se aplicaría cuando hay menos de estas 3 características presentes.
La hiperplasia seudoepiteliomatosa fue un hallazgo bastante constante en nuestra serie presente hasta en el 58,82 % de los casos y que se repite en otras series publicadas. Este hecho parece importante a la hora de hacer el diagnóstico diferencial con el carcinoma espinocelular, con el que a menudo se confunde17 (fig. 4).
En conclusión, podemos decir que el TCG es un tumor raro, que no se sospecha clínicamente salvo cuando se localiza en la lengua, y que tiene un origen aparentemente neural a partir de las células de Schwann, salvo la variedad de TCG no neural de la que no hemos hallado ningún caso en nuestra serie. Son raros los casos familiares, congénitos y de afectación múltiple y cabe recalcar el buen pronóstico de este tumor, no detectándose recurrencias en nuestra serie tras la exéresis quirúrgica completa. La presencia de «CPO de Milian» recientemente descrita podría ser un hallazgo histológico adicional en el diagnóstico de este tumor por su elevada prevalencia en las muestras analizadas. Parece ser que conforme aumenta la edad del tumor se incrementaría el número de CPO. Estos serían el resultado de la progresiva acumulación de los gránulos en el interior de los lisosomas que por su madurez han perdido mitocondrias y retículo endoplásmico, por lo que las células tienen más tendencia para la segregación focal de su material citoplásmico, originando así estos CPO10.
Nuestra serie confirma las características descritas hasta el momento para el TCG, salvo algunas peculiaridades como son la alta prevalencia en niños y adultos jóvenes, y confirma la presencia de CPO en el examen microscópico. Nos parece interesante seguir investigando los orígenes del TCG, ya que, aunque en nuestra serie la positividad para la proteína S-100 apoya un origen neural, existen series publicadas donde no se confirma este origen15,16.
Nos parece apropiado el examen físico completo a todo paciente que presente algún TCG para descartar la presencia de tumores múltiples asociados y una posible afectación visceral. En los niños este examen físico adquiere mayor importancia por la asociación posible, aunque poco frecuente, con anomalías sistémicas musculoesqueléticas, cardiovasculares y neurológicas que pueden comprometer la vida de estos pacientes8,9.
Conflicto de intereses
Declaramos no tener ningún conflicto de intereses.


