







Caso clínico
Niña de 4 meses de edad nacida a pretérmino, que desde el nacimiento presentaba una piel extremadamente seca de aspecto ictiosiforme, junto con alteraciones en el pelo y las uñas. En la exploración se apreciaba fascie de progeria junto a una fragilidad capilar importante (figs. 1 y 2). La piel tenía un aspecto ictiosiforme que también afectaba a palmas y plantas (fig. 3). Se acompañaba de retraso en el crecimiento, con peso, longitud y perímetro craneal por debajo del P3, existiendo una discordancia evidente entre longitud-peso. El desarrollo psíquico era normal. A lo largo de los controles la madre refería problemas de fotosensibilidad y clínicamente era evidente la presencia de lentigos.
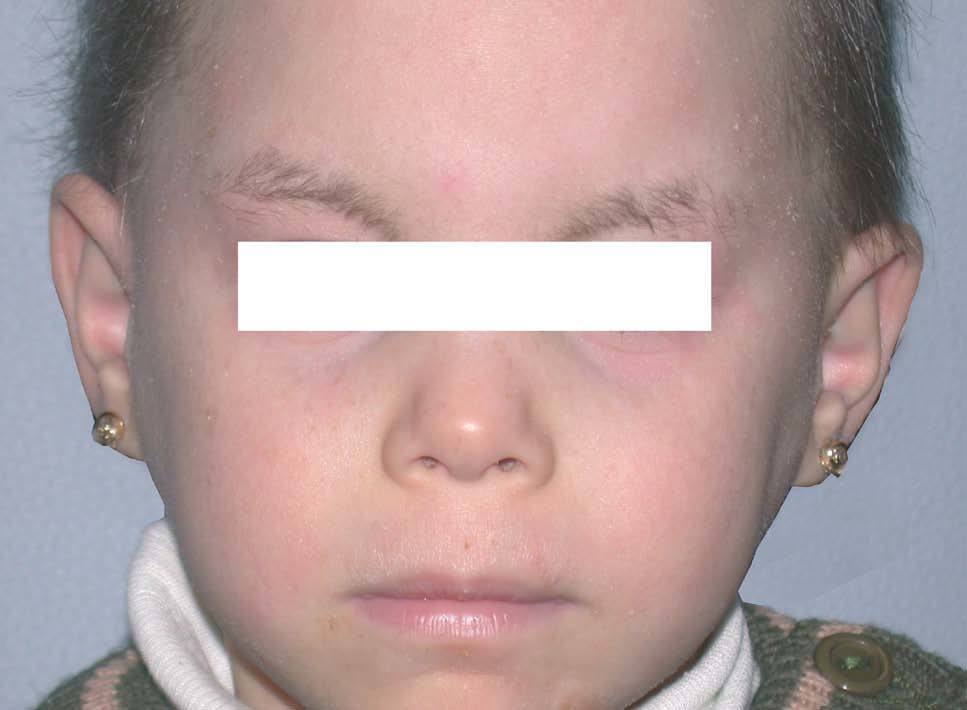
Figura 1. Fascies de progeria.
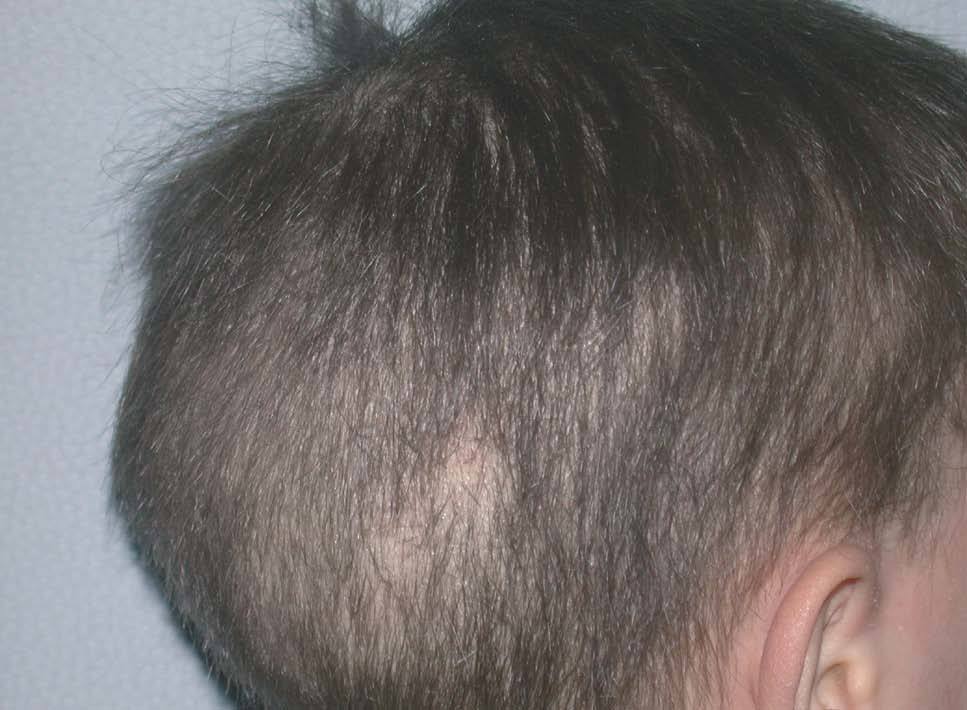
Figura 2. Aspecto capilar característico, con fragilidad importante.
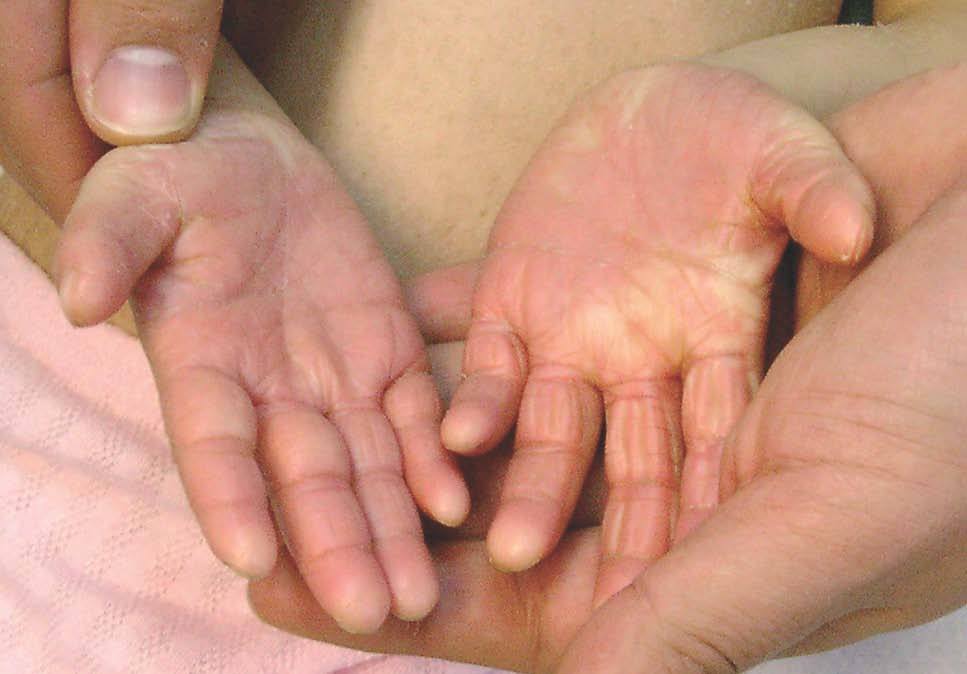
Figura 3. Piel de aspecto ictiosiforme con afectación de palmas y plantas.
Entre los antecedentes personales caben destacar patologías poco frecuentes en esta edad, como una enterocolitis por Campylobacter y dos procesos neumónicos. Debido a estas alteraciones se realizaron precozmente estudios citogenéticos e inmunitarios que resultaron normales.
Las exploraciones complementarias mostraron una anemia microcítica hipocroma catalogada como rasgo talasémico, por el aumento del número de hematíes asociado a la disminución del volumen corpuscular medio (VCM) y a los antecedentes familiares.
Se tomaron muestras para un examen tricológico completo. El tricograma mostró una trichosquisis por trichorrhexis nodosa y presencia esporádica de pili torti (fig. 4). El estudio de componentes demostró un déficit leve de azufre en pelos de la región fronto-parietal, más evidente (> 50 %) en los de la región occipital (fig. 5).
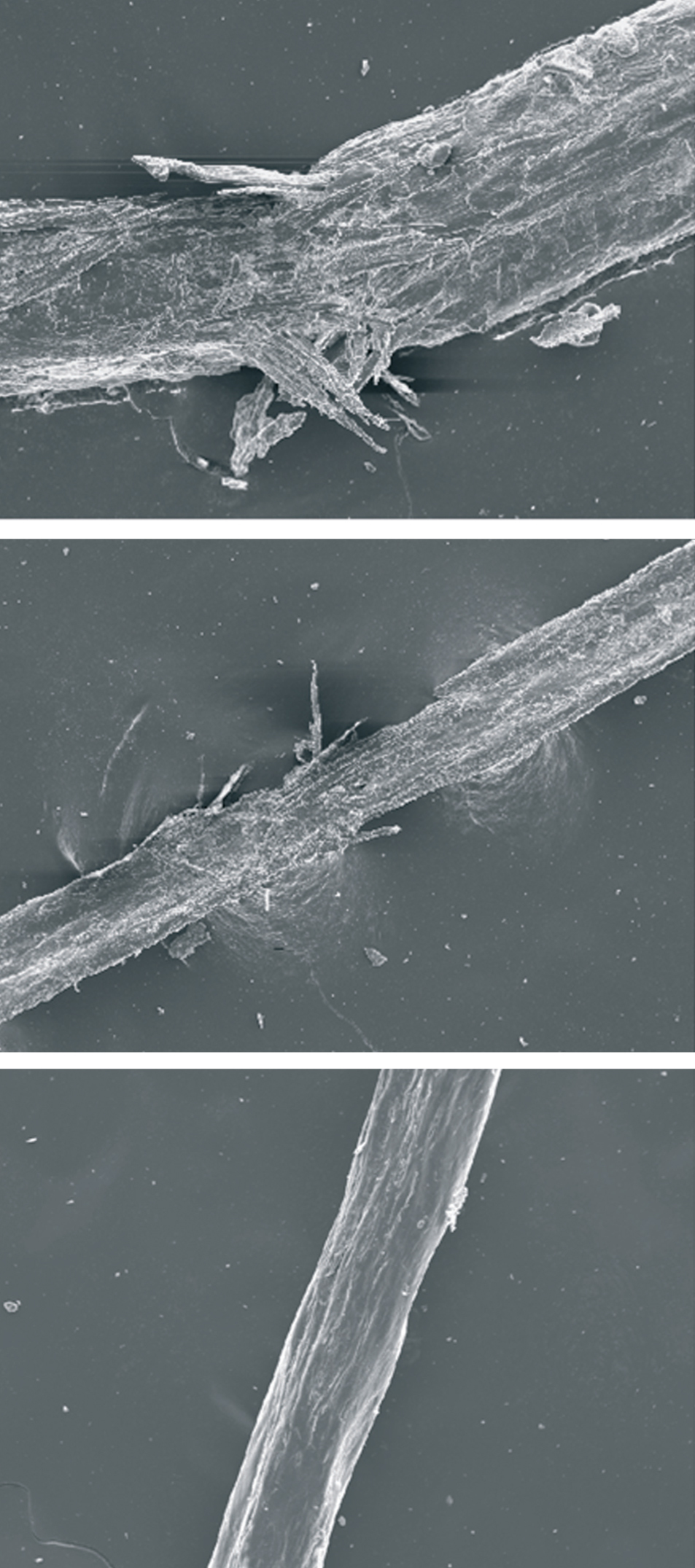
Figura 4. Tricosquisis (tricorrexis nodosa) y pili torti en el tricograma.
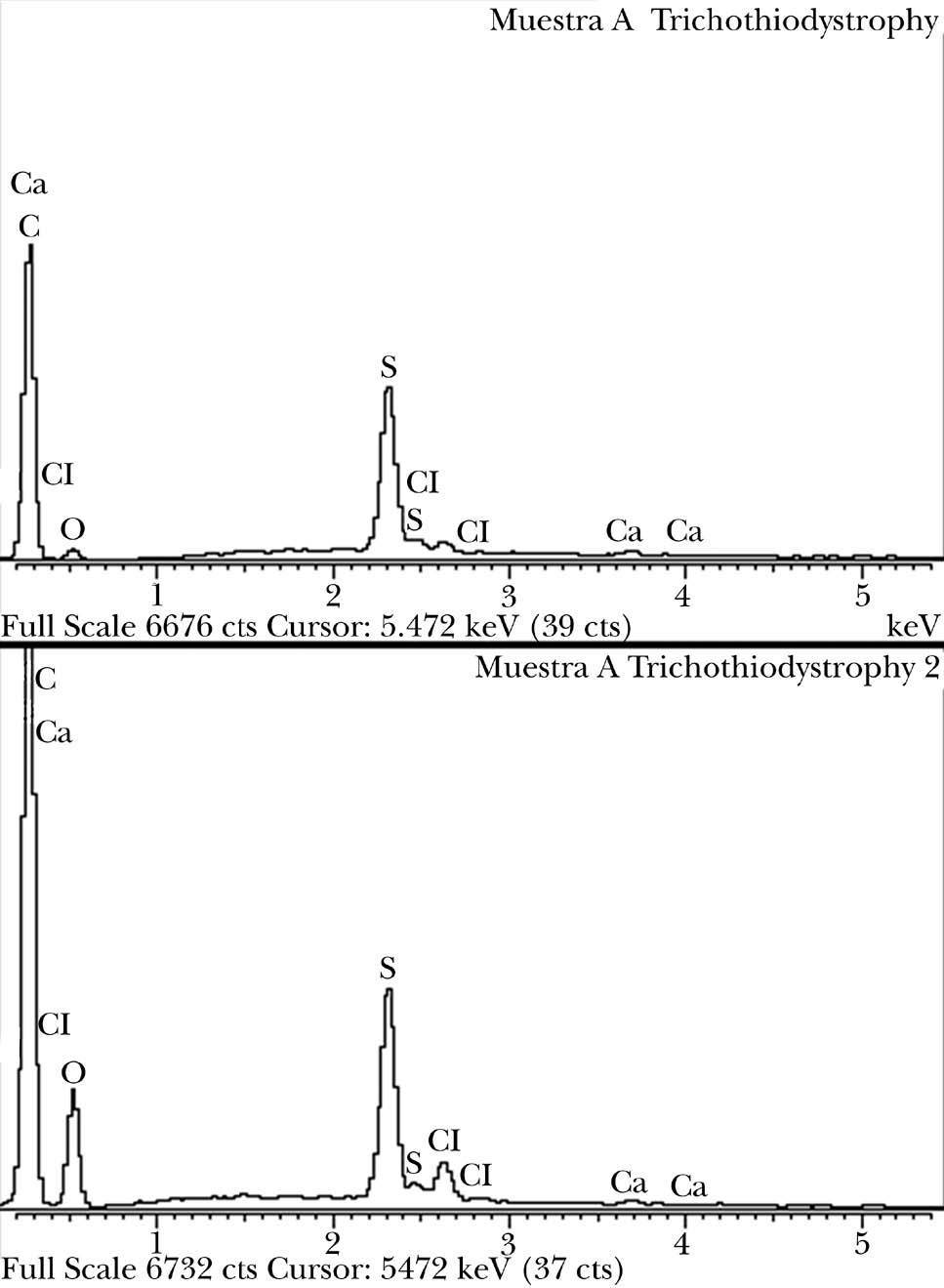
Figura 5. Déficit de azufre (> 50 %).
Dadas las manifestaciones clínicas y complementarias se llegó al diagnóstico de tricotiodistrofia, que al asociar la eritrodermia ictiosiforme con alteraciones en el pelo y retraso físico se catalogó inicialmente como síndrome Tay. La posterior aparición de fotosensibilidad en la evolución del cuadro y la presencia de múltiples lesiones pigmentarias faciales (lentigos) nos hizo sospechar finalmente de que se trataba de un PIBIDS.
Discusión
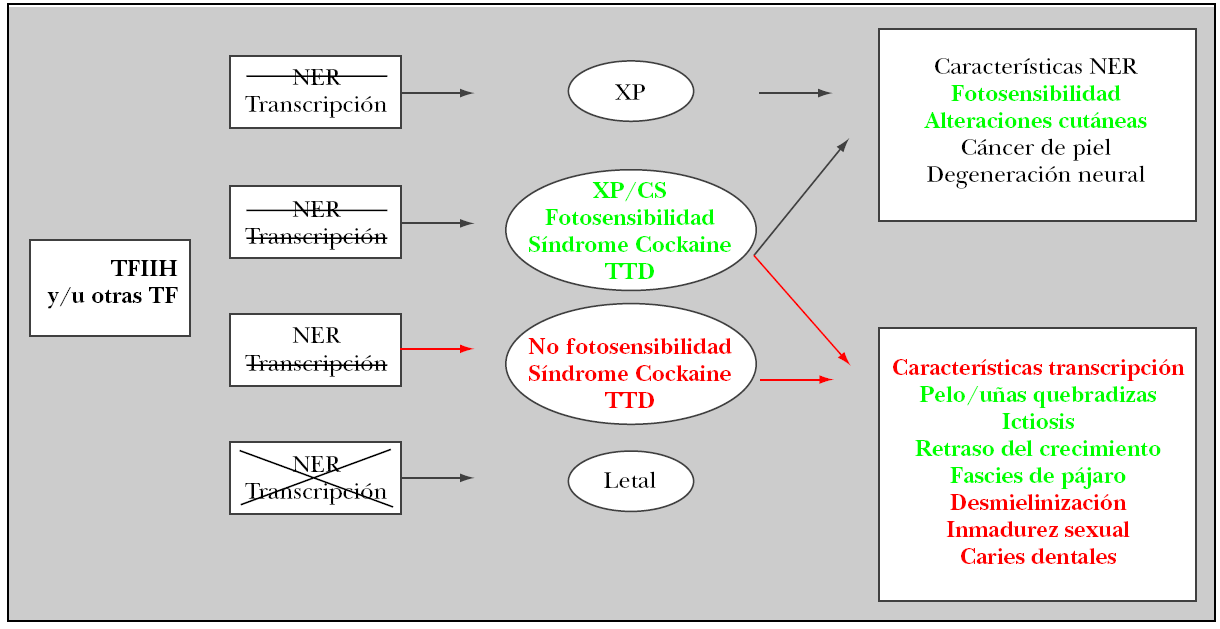
El término tricotiodistrofia deriva del griego tricho: pelo, thio: azufre, dis: alteración, trophe: forma. El origen de este desorden se encuentra en el déficit de azufre que existe en tejidos derivados del neuroectodermo. La fotosensibilidad observada deriva de un defecto en el sistema de reparación de escisión nucleótida (NER)1,2 que se encarga de la reparación del ADN. El defecto en alguno de los 11 genes que determinan la afectación en la función del general transcription factor IIH (TFIIH), conlleva padecer un alto riesgo de cáncer cutáneo por hipersensibilidad cutánea a la radiación ultravioleta (UV). Según se encuentre afectada la función del complejo NER, de la transcripción o ambas, se observarán una serie de características clínicas que componen síndromes diferentes (fig. 6). Son tres las entidades más frecuentemente relacionadas con la alteración de este sistema3,4: xeroderma pigmentosum, síndrome de Cockayne y tricotiodistrofia (PIBIDS).
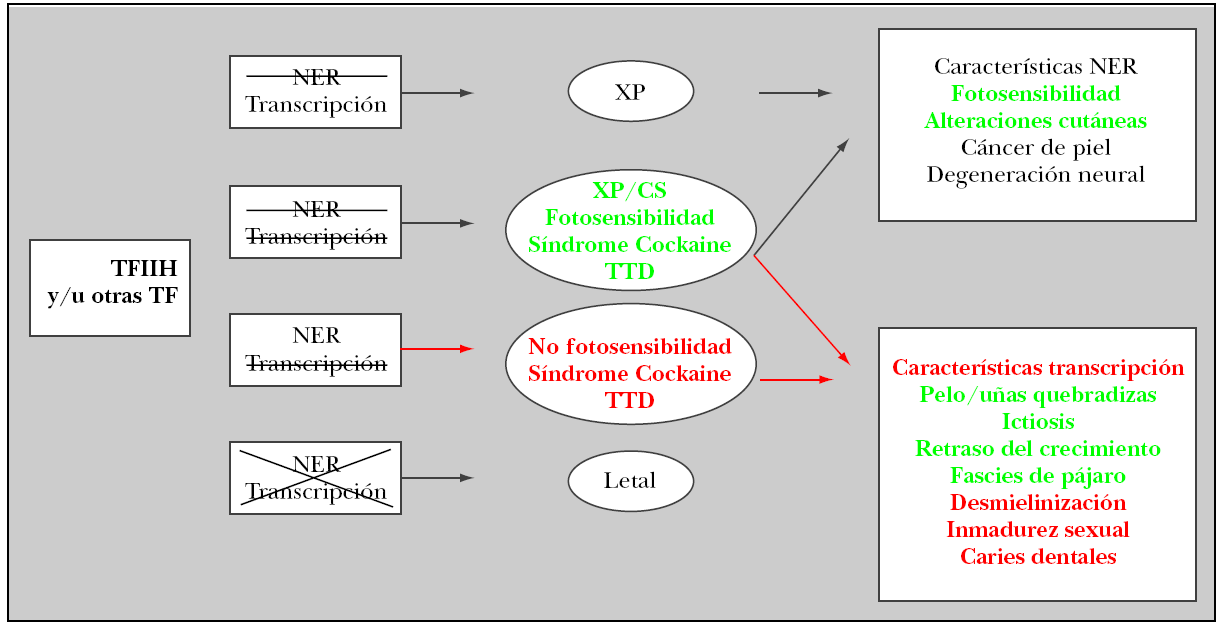
Figura 6. Sistema de reparación de escisión nucleótida (NER). Genes implicados y manifestaciones clínicas. Tachado = déficit parcial; cruz = déficit total.
El término fue adoptado inicialmente por Price entre los años 1979-1980, y se basó en una serie de casos a los que sumó otros descritos por Pollit, Jenner y Davies (1968). Estos enfermos asociaban retraso mental y físico, así como trichorrhexis nodosa. Posteriormente, Brown et al (1970) describieron el defecto específico que se produce en el pelo, la trichosquisis, así como una birrefringencia alterna, y la determinación de un contenido bajo en azufre.
Tay (1971) describió la asociación con eritrodermia ictiosiforme, retraso mental y físico, denominándolo síndrome Tay. Jorizzo et al (1982) sugieren el término IBIDS, que asocia ictiosis. Posteriormente se describen signos que completan el cuadro, así Chapmann (1988) propone SIBIDS que asocia además osteosclerosis, y Crovato, Borrone y Rebora (1983) proponen el término PIBIDS, que incluye la fotosensibilidad5 (tabla 1).

Clínicamente se observa un pelo característicamente quebradizo, desarrollo de fotosensibilidad hasta en un 50 % de los casos, debida a una deficiencia en la reparación del ADN, prácticamente indistinguible del producido en el XP tipo D6, aunque sin evidencia de aumento del cáncer cutáneo en esta entidad. Existen numerosas manifestaciones asociadas descritas en forma de casos clínicos aislados7 que resumimos en la tabla 2 junto a las formas más frecuentes.

Desde el punto de vista diagnóstico las alteraciones del pelo son los únicos cambios diagnósticos (y obligatorios) que identifican el déficit de azufre. Los cambios macroscópicos se aprecian en las regiones frontal y occipital, y los microscópicos en la región occipital. Así, en el microscopio óptico se observan característicamente imágenes de trichosquisis, pero pueden observarse otras distrofias pilosas como trichorrhexis nodosa atípica o alteraciones pili torti-like. En el microscopio de luz polarizada se aprecia un patrón tiger-tail8. Por otro lado, el análisis del contenido de azufre debe encontrarse en cifras inferiores al 50 %.
Desde el punto de vista terapéutico cabe destacar el avance en el diagnóstico prenatal de nuevos casos en madres embarazadas que presenten algún hijo afecto, basándose en la medida de defectos en la síntesis de ADN inducidos por UV en células cultivadas de líquido amniótico, que puede llevar a los padres a tomar la decisión de detener el embarazo en algunos casos9.
Conflicto de intereses
Declaramos no tener ningún conflicto de intereses.
Correspondencia:
Rafael Jiménez-Puya.
Servicio de Dermatología. Hospital Universitario Reina Sofía.
Avda. Menéndez Pidal, s/n. 14004 Córdoba.
JIMPUYA@terra.es
Aceptado el 16 de noviembre de 2006.


