


A 30-year-old female was seen in the outpatient clinic with recurrent bruising that appeared spontaneously, or after minimal trauma; these were barely painful and would disappear on their own after a few weeks leaving a greenish discoloration. She had a personal history of migraine and myopia, her mother had a history of surgery for retinal detachments in several occasions, and, since her father had a diagnosis of Von Willebrand's disease, she had already been evaluated by hematology, who ruled out a coagulation disorder after finding normal coagulation tests and platelet aggregation curve. Nevertheless, coagulation function was reassessed finding normal PT, PTT, platelet aggregation curve, VWF: Ag and hemogram, although a slightly prolonged bleeding time was found (13/9min).
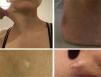
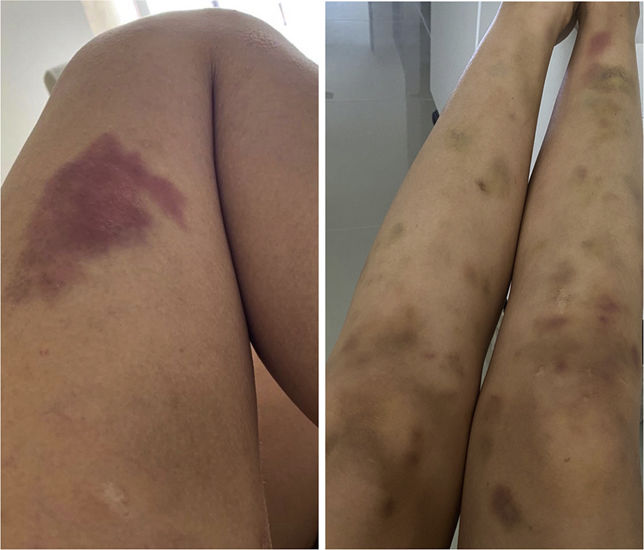
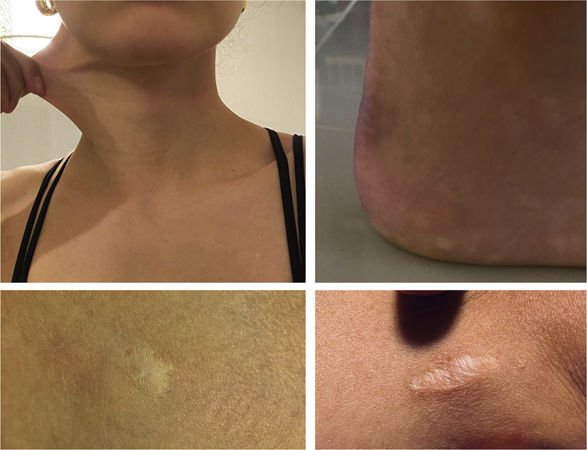
Physical examinationThe initial examination revealed several ecchymoses of different sizes predominantly in the lower extremities (Fig. 1). Her skin was velvety in appearance and to the touch, she had some atrophic scars on her legs and arms, piezogenic papules and hyperelasticity of the skin (Fig. 2). With these findings, joint mobility was assessed, scoring 7/9 in the Beighton scale (Fig. 3); based on these clinical findings, a diagnosis of hypermobile Ehlers-Danlos Syndrome (hEDS) was proposed.
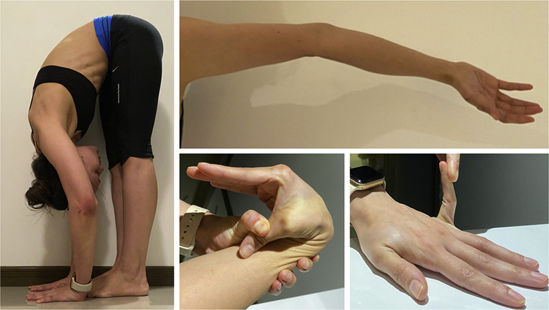
Assessment of joint hypermobility with the Beighton score: Ability to flex spine placing palms to floor without bending knees (Left) – 1 point; Active hyperextension of elbow >10° (Superior right) – 1 point each side; Active hyperextension of knee >10° – was not present; Passive apposition of thumb to forearm (Bottom middle) – 1 point each side; Passive hyperextension of fifth metacarpal-phalangeal joint >90° (Bottom right) – 1 point each side.
What is the diagnosis?
Diagnosis and evolutionThe patient was referred to genetics, who considered ruling out classical EDS and ordered a genetic panel and complementary tests. A mild mitral prolapse was identified through M-mode echocardiogram (posterior mitral leaflet 2mm above the annular plane). MRI and X-rays of the dorsolumbar spine and knees demonstrated mild scoliosis with facet joint stenosis, bilateral patellofemoral malalignment, and grade 1 chondromalacia patellae. The ophthalmologic evaluation found bilateral lattice degeneration of the retina with a small peripheral tear in the left eye, which was corrected through photocoagulation.
The EDS genetic panel was negative but diagnostic criteria for hEDS were met by the patient (Table 1. Supplementary data), and also by her mother. Paraclinical findings allowed starting an opportune multidisciplinary approach to attenuate further deterioration. Treatment with ascorbic acid 2g daily was started, the patient referred decreased bruising.
DiscussionEDS encloses a broad group of genetic disorders of the connective tissue; according to the latest classification, there are 13 subtypes, being hypermobile the most frequent one with an estimated prevalence of 1:5000.1 hEDS is inherited in an autosomal dominant pattern, it is characterized by generalized joint hypermobility predisposing to instability and early degenerative joint disease, skin hyperextensibility that is milder than in other EDS subtypes, and easy bruising. It may be associated with mitral valve prolapse, migraines, postural orthostatic tachycardia syndrome (POTS), ocular alterations (including myopia and predisposition to retinal detachments), chronic pain, and psychologic disturbances.2–4
A genetic mutation hasn’t been identified yet, hence the diagnosis remains clinical and based on the criteria established by the International Consortium on Ehlers-Danlos Syndrome & Related Disorders in association with the Ehlers-Danlos society1; three criteria must be met: The presence of generalized joint hypermobility; systemic manifestations, family history, and/or musculoskeletal complications; and the exclusion of alternate diagnosis (Table 1).
Given the broad compromise, patients require multidisciplinary management aiming to prevent the complications that might present, and should be tailored to the particular manifestations of each patient; there is no standardized approach for soft tissue fragility and its manifestations, but the use of high doses of ascorbic acid (1–4g daily) may improve wound healing and decrease easy bruising.5 It is also advisable, when practicing procedures in these patients, to consider longer times for suture removal, up to 5 days later than in non-EDS patients.6
Ehlers-Danlos syndrome is an entity with a variable presentation that usually has cutaneous manifestations, these may be the leading cause of consultation, so the dermatologist must be aware and alert to the identification of the different signs suggestive of the disease. In this case, the presence of ecchymoses led to the diagnosis and allowed the opportune identification and management of subclinical multisystemic compromise.
Conflict of interestsThe authors declare that they have no conflict of interest.
The following are the supplementary data to this article:



