


The image corresponds to a 38-year-old man who was admitted to the neurology department with a 3-month history of weakness in both legs and difficulty walking (Fig. 1). Physical examination revealed leg weakness, tandem gait difficulty, and universal areflexia. During admission, the patient was observed to have hepatosplenomegaly, multiple enlarged lymph nodes, and thrombocytosis. The neurophysiological study revealed predominantly demyelinating sensorimotor polyneuropathy. The initial diagnosis was chronic inflammatory demyelinating polyneuropathy, and the patient received treatment with corticosteroids and intravenous immunoglobulin boluses. Three months later, owing to a loss of visual acuity, he was assessed in ophthalmology, where he was diagnosed with incipient bilateral papilledema. He also underwent lumbar puncture, with normal opening pressure and markedly increased cerebrospinal fluid proteins. In addition, he had subclinical hypothyroidism and slightly increased prolactin values.
Physical Examination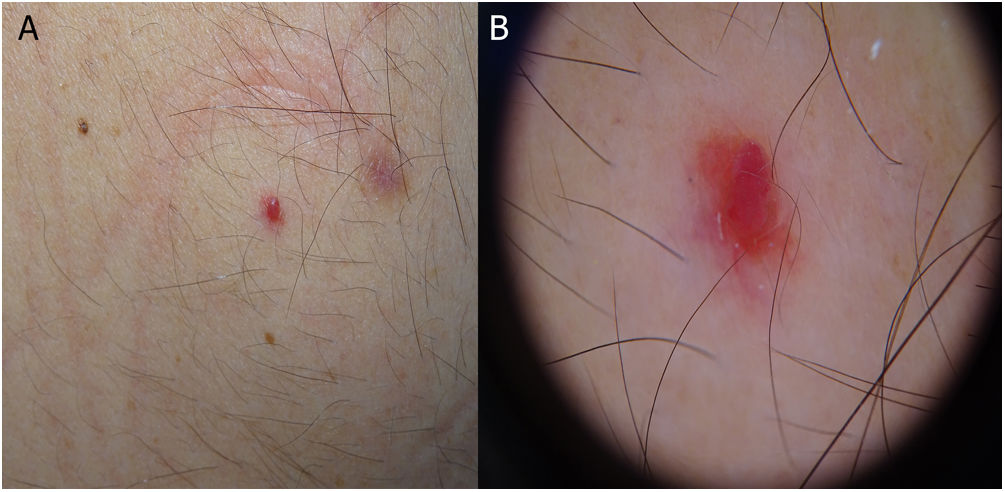
Five recent lesions were observed on the skin. Examination revealed red dome-shaped papules that were clinically and dermoscopically compatible with cherry angiomas (Fig. 1).
HistopathologyExamination of a punch biopsy specimen from the largest lesion indicated glomeruloid hemangiomas (Fig. 2).
 Hematoxylin-eosin, ×5. (B) Hematoxylin-eosin, ×10. (C and D) Hematoxylin-eosin, ×40.")
What is your diagnosis?
DiagnosisPOEMS syndrome.
Clinical Course and TreatmentExtension of the diagnostic study revealed increased vascular endothelial growth factor (VEGF) and absence of clonal plasma cells, leading to a diagnosis of atypical, or incomplete, POEMS syndrome. Four months later, analysis revealed the presence of a low-intensity monoclonal immunoglobulin G κ component in blood, with markedly increased VEGF values (2407pg/mL). Therefore, a definitive diagnosis of POEMS syndrome was made based on the presence of the 2 mandatory components (polyneuropathy and a monoclonal component), a major component (increased VEGF), and 5 minor components (hepatosplenomegaly, endocrinopathy, skin lesions, papilledema, and thrombocytosis). Consequently, the patient underwent autologous stem cell transplantation, with a favorable initial response.
CommentPOEMS syndrome is a rare paraneoplastic syndrome caused by underlying monoclonal plasma cell dyscrasia. The acronym was coined by Bardwick in 1980 to refer to several of the characteristics of the syndrome: polyradiculoneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell dyscrasia, and skin changes. To date, VEGF is the cytokine that best correlates with disease activity.1 The cutoff values for POEMS syndrome are 200pg/mL in plasma (specificity of 95% and sensitivity of 68%) and 1920pg/mL in serum (specificity of 98% and sensitivity of 73%).1,2 Diagnosis is based on the criteria of Dispenzieri,1 which require 2 mandatory criteria to be met, namely, 1 major and 1 minor.
Skin manifestations affect 70%–90% of patients and include hyperpigmentation, acrocyanosis, facial plethora, hemangiomas, hypertrichosis, and scleroderma-like skin changes.1,3 Glomeruloid hemangioma was first described by Chan et al. in 1990, and while it is uncommon, it constitutes the most specific skin manifestation of POEMS syndrome, with a strong association between both.4 It is believed to be a proliferative reaction to angiogenic stimuli (VEGF, interleukin 1, and tumor necrosis factor) and usually appears abruptly as multiple violaceous dome-shaped papules with a vascular appearance on the trunk and proximal area of the extremities. Sudden onset over a few days or weeks helps to establish an association between glomeruloid hemangioma and POEMS syndrome, since a slower onset may be observed in patients with no associated symptoms.3 Furthermore, it is important to stress that glomeruloid hemangiomas can precede full development of the disease by some years, thus providing an early sign of POEMS syndrome.5
Atypical, or incomplete, POEMS syndrome4,6 is characterized by the absence of one of the mandatory criteria. This condition requires close follow-up, since progression to the complete form has been reported, as in the case we present.
Treatment ranges from radiotherapy to systemic approaches similar to those used in multiple myeloma.
Conflicts of InterestThe authors declare that they have no conflicts of interest.


