



Mycosis fungoides (MF) is the most common primary cutaneous T-cell lymphoma. The clinical course of the disease is typically characterized by progression from a nonspecific phase of erythematous macules to the appearance of plaques and ultimately, in some patients, tumors. However, numerous clinical and histopathologic variants of MF with specific therapeutic and prognostic implications have been described in recent decades. Clarification of the differential diagnosis can be frustrated by the wide range of clinical manifestations and histopathologic patterns of cutaneous infiltration, particularly in the early phases of the disease. In this paper, we review the main clinical, histopathologic, and immunohistochemical characteristics of the variants of MF described in the literature in order to facilitate early diagnosis of the disease.
La micosis fungoide (MF) es el linfoma primario cutáneo de células T más frecuente. La evolución clínica clásica de la enfermedad se caracteriza por la progresión desde una fase inespecífica de máculas eritematosas a la aparición de placas y, finalmente, tumores en algunos pacientes. Sin embargo, a lo largo de las últimas décadas se han descrito numerosas variantes de MF, tanto desde el punto de vista clínico como histopatológico, con implicaciones terapéuticas y pronósticas específicas. El diagnóstico diferencial se ve dificultado así ante el amplio abanico de manifestaciones clínicas y patrones histopatológicos de infiltración cutánea, especialmente en fases precoces de la enfermedad. Este artículo revisa las principales características clínicas, histopatológicas e inmunohistoquímicas que definen las distintas variantes de MF descritas en la literatura con el objetivo de facilitar el diagnóstico temprano de MF.
Mycosis fungoides (MF) is the most common primary T-cell cutaneous lymphoma and accounts for almost 50% of all primary cutaneous lymphomas.1 Described for the first time in 1806 by the French dermatologist Jean Louis Alibert, classic MF starts with a nonspecific phase consisting of erythematous macules that can last for years. In subsequent phases, patients develop plaques and, in some cases, tumors. The name mycosis fungoides refers to the mushroom-like appearance of the tumors. Enlarged lymph nodes, visceral involvement, and transformation to large-cell lymphoma are less common findings that are typically seen in advanced stages of the disease. Numerous clinical and histopathologic variants of MF have been described in recent decades. Although some of these variants have been reported as isolated cases, others have greater clinical relevance due to their relative frequency and their therapeutic and prognostic implications. The wide range of clinical and pathological presentations of early-stage MF requires a broad differential diagnosis as early lesions can mimic many of the patterns seen in cutaneous inflammatory disorders.
In this article, we review the main clinical, histopathologic, and immunohistochemical characteristics that help to establish a correct diagnosis of MF, particularly in the early stages of disease. We also describe the distinctive clinical and pathological features that define the different variants of MF.
Classic MFMF has traditionally been defined as a 3-stage disorder characterized by the progressive appearance of patches, plaques, and tumors. Not all patients, however, pass through these 3 stages. Some remain in the plaque stage, showing no signs of disease progression, while others develop patches, plaques, and tumors as a presenting form of the disease.
Patch-Stage MFPatch-stage MF is clinically characterized by the presence of asymmetric, irregular, erythematous macules and patches occasionally associated with atrophy and/or telangiectasia.2 The lesions are generally asymptomatic or mildly pruritic and disappear spontaneously, without leaving residual lesions (Fig. 1A). Patch-stage MF can last for years, with no signs of progression. It is not associated with worse outcomes than other forms of MF and patients have a similar life expectancy to the general population.3
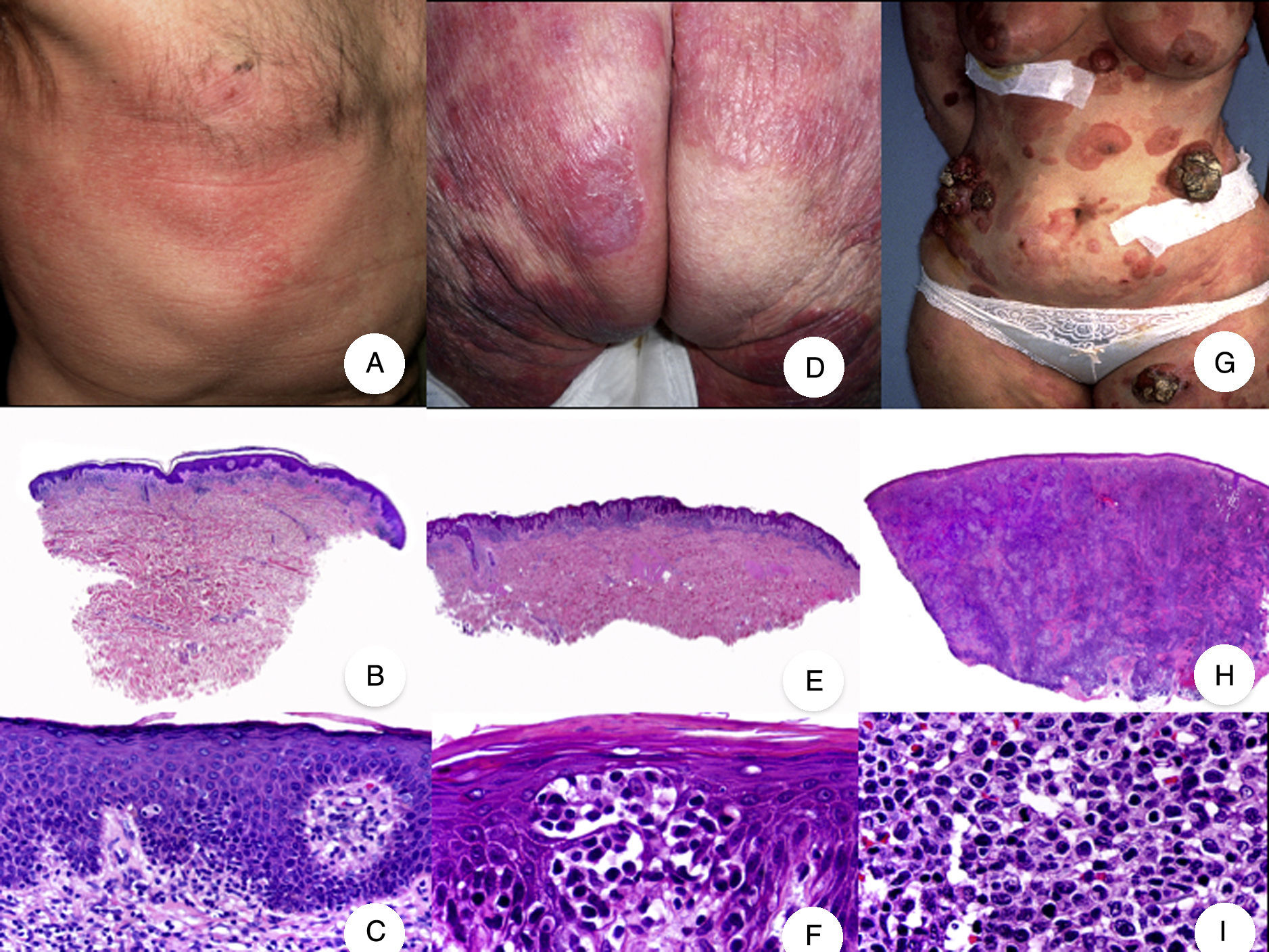
. A, Photograph of patch-stage MF lesions. B, C, Typical histopathologic findings in patch-stage MF showing an epidermotropic infiltrate composed of atypical lymphocytes in the papillary dermis. D, Photograph of plaque-stage MF lesions. E,F, Typical histopathologic findings in plaque-stage MF; they are similar to those seen in patch-stage MF, but there is a denser lichenoid lymphocytic infiltrate with a band-like distribution in the superficial dermis, together with marked epidermotropism and Pautrier microabscesses. G, Photograph of tumor-stage MF. H, I, Characteristic histopathologic features in tumor-stage MF, including a diffuse lymphocytic infiltrate of large, irregular lymphocytes occupying the full dermis.")
Classic mycosis fungoides (MF). A, Photograph of patch-stage MF lesions. B, C, Typical histopathologic findings in patch-stage MF showing an epidermotropic infiltrate composed of atypical lymphocytes in the papillary dermis. D, Photograph of plaque-stage MF lesions. E,F, Typical histopathologic findings in plaque-stage MF; they are similar to those seen in patch-stage MF, but there is a denser lichenoid lymphocytic infiltrate with a band-like distribution in the superficial dermis, together with marked epidermotropism and Pautrier microabscesses. G, Photograph of tumor-stage MF. H, I, Characteristic histopathologic features in tumor-stage MF, including a diffuse lymphocytic infiltrate of large, irregular lymphocytes occupying the full dermis.
The histopathologic features of patch-stage MF include scarce lymphocytes scattered through the basal layers of the epidermis, accompanied by focal parakeratosis, papillary dermal fibrosis, and a larger population of lymphocytes arranged in a band-like pattern along the dermal-epidermal junction. Additional findings include a predominantly lymphocytic perivascular, periadnexal, or subepidermal infiltrate with eosinophils and some plasma cells (Fig. 1B and C). Intraepidermal lymphocytes are generally larger and more pleomorphic than lymphocytes found in the superficial dermis, and on occasions they are surrounded by a pale cytoplasmic halo. These morphologic features are highly indicative of MF, but they are not consistent, and repeat skin biopsies over time are often necessary to establish a definitive diagnosis of patch-stage MF.4
Plaque-Stage MFPlaque-stage MF is characterized by well-demarcated erythematous or dark brown plaques that are often pruritic and accompanied by scaling. They generally affect a large area of skin (Fig. 1D). Plaques and characteristic macules from earlier stages frequently coexist in the same or different areas.5,6
Plaque-stage MF has similar histopathologic features to patch-stage MF but it has a denser band-like lichenoid lymphocytic infiltrate in the superficial dermis. This infiltrate is primarily composed of small and medium-sized lymphocytes with pleomorphic hyperchromatic nuclei and numerous convolutions that lend it a cerebriform appearance (Fig. 1E and F). Epidermotropism is frequently more pronounced in plaque-stage than in patch-stage MF and is characterized by the presence of atypical lymphocytes in the epidermis that occur in isolation or form collections known as Pautrier microabscesses. Other common findings include epidermal hyperplasia, papillary dermal fibrosis, and some eosinophils and plasma cells in the accompanying inflammatory infiltrate.
Tumor-Stage MFSkin tumors are the key clinical feature of tumor-stage MF and they frequently coexist with patches and plaques (Fig. 1G). The absence of patches and plaques should lead to a reassessment of diagnosis and the inclusion of other more aggressive non-MF cutaneous lymphomas in the differential diagnosis. MF tumors are characterized by significant vertical growth, giving rise to smooth reddish-brown or bluish-red nodules that can reach a size of several centimeters and become ulcerated or infected.7
Histopathologic findings include a diffuse nodular lymphocytic infiltrate formed by large pleomorphic lymphocytes with hyperchromatic nuclei and prominent nucleoli occupying the full thickness of the dermis and possibly extending into the subcutaneous tissue (Fig. 1H and I). The cells in the infiltrate frequently have a high proliferative index and typical and atypical mitotic figures are abundant. Pautrier microabscesses and epidermotropism are normally absent in the tumor stage of MF.
Diagnosing MFPathologic diagnosis of early-stage MF is challenging not only because of the subtle nature of the histopathologic findings, but also because of overlapping with features seen in other inflammatory skin disorders. This is further complicated by the absence of certain characteristic clinical and histopathologic findings in early MF. Pautrier microabscesses, for instance, are observed in less than 25% of cases, atypical lymphocytes are found in less than 10% of cases, and epidermotropism, a hallmark feature of MF, may be absent in up to 4% of cases.1,8
In 2005, the International Society for Cutaneous Lymphomas developed a diagnostic algorithm combining clinical, histopathologic, immunohistochemical, and molecular criteria to facilitate the diagnosis of early-stage MF (Table 1)9; the algorithm supports the use of serial skin biopsies in equivocal cases.10
Algorithm for Diagnosing Early-Stage Mycosis Fungoides.
| Criteria | Scorea |
|---|---|
| Clinical criteria Main criterion Persistent and/or progressive patches and plaques Additional criteria Nonphotoexposed areas Variable size and shape Poikiloderma | 2 points for main criterion plus 2 additional criteria 1 point for main criterion plus 1 additional criterion |
| Histopathologic criteria Main criterion Superficial lymphocytic infiltrate Additional criteria Epidermotropism without spongiosis Atypical lymphocytes (cells with large, hyperchromatic, and irregular or cerebriform nuclei) | 2 points for main criterion plus 2 additional criteria 1 point for main criterion plus 1 additional criterion |
| Molecular biology criteria Monoclonal T-cell receptor gene rearrangement | 1 point for clonality |
| Immunopathologic criteria <50% CD2+, CD3+, and/or CD5+T cells <10% CD7+T cells Dermal/epidermal discordance in expression of CD2, CD3, CD5, or CD7b | 1 point for at least 1 criterion |
Immunohistochemical analysis of neoplastic T cells, combined with clinical and histopathologic findings, can aid diagnosis. Neoplastic cells in MF are typically CD4+, with variable loss of other T-cell markers, such as CD2, CD3, CD5, CD7, and CD26. Within this group, loss of CD7, followed by CD5, is the most common finding. A CD8+ T-cell phenotype may be detected in up to 20% of cases. Monoclonal T-cell receptor (TCR) gene rearrangement may also point to a diagnosis of early-stage MF. Unfortunately, neither TCR gene rearrangement nor loss of CD7 expression permits a definitive diagnosis, as these nonspecific findings are also seen in other benign inflammatory skin conditions and are probably a result of chronic T-cell stimulation.11
Despite the lack of specific cellular and molecular markers for confirming early-stage MF, 19 different genes were recently found to be significantly upregulated in early MF compared with other chronic inflammatory disorders.12 Two of these genes—programmed cell death protein 1 gene (PDCD1) and the thymocyte selection-associated high mobility group box gene (TOX)—are of particular interest due to their capacity to discriminate between early MF and benign inflammatory conditions. TOX appears to be both a sensitive and specific immunohistochemical marker for the early diagnosis of MF and displays an intense diffuse nuclear staining pattern that is not seen in other inflammatory skin disorders. TOX-positive cells have atypical nuclei and are seen in both the epidermis and papillary dermis of skin biopsy specimens from patients with early-stage MF, as well in Pautrier microabscesses. A close correlation has been found between TOX staining and neoplastic cells in MF.12
Clinicopathologic Variants of MFThe clinicopathologic variants of MF fall along a spectrum ranging from clinical variants, which have distinctive clinical but similar histopathologic features to classic MF, to clinicopathologic variants, which have distinctive clinical and histopathologic features, to histopathologic variants, which can only be distinguished from classic MF by biopsy (Table 2). Within this broad group, only folliculotropic MF, pagetoid reticulosis, and granulomatous slack skin were included in the latest international classification of cutaneous lymphomas published by the World Health Organization (WHO) in 2016. In this review, however, we describe the full spectrum of clinicopathologic variants, as familiarity with these will help to narrow the differential diagnosis for both dermatologists and pathodermatologists.
Clinical, Clinicopathologic, and Histologic Variants of Mycosis Fungoides.
| Clinical Variants |
|---|
| Hypopigmented mycosis fungoides Erythrodermic mycosis fungoides Ichthyosiform mycosis fungoides Mycosis fungoides palmaris et plantaris Papillomatous mycosis fungoides Papular mycosis fungoides Solitary or unilesional mycosis fungoides Invisible mycosis fungoides |
| Clinicopathologic variants |
|---|
| Folliculotropic mycosis fungoides Mycosis fungoides with eruptive infundibular cysts Syringotropic mycosis fungoides Granulomatous slack skin Pagetoid reticulosis or Woringer-Kolopp disease Poikilodermal mycosis fungoides (poikiloderma vasculare atrophicans) Bullous mycosis fungoides and dyshidrotic mycosis fungoides Anetodermic mycosis fungoides Hyperpigmented mycosis fungoides Purpuric mycosis fungoides Pustular mycosis fungoides Verrucous mycosis fungoides |
| Histopathologic variants |
|---|
| Granulomatous mycosis fungoides Interstitial mycosis fungoides Mycosis fungoides with large-cell transformation |
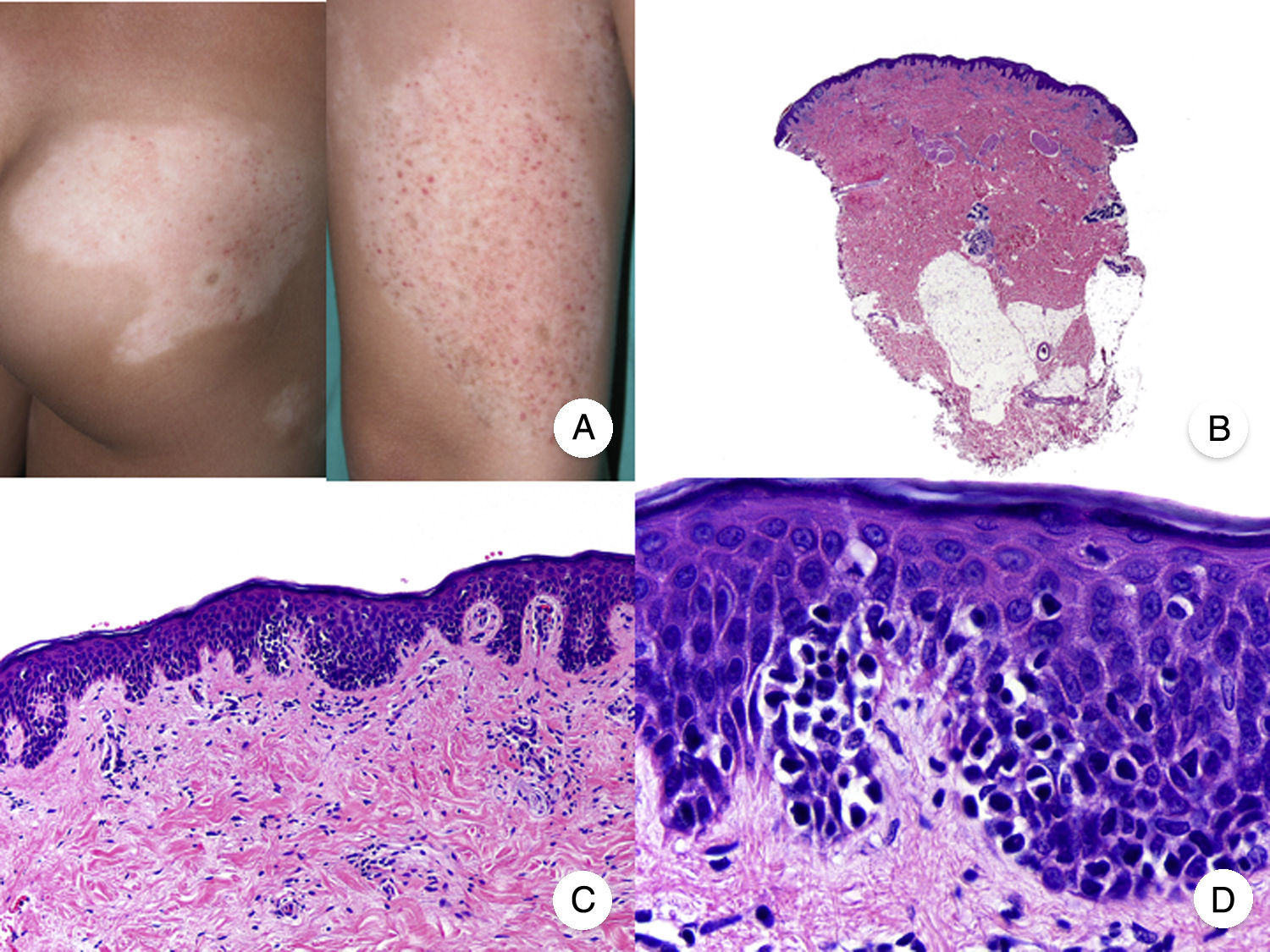
Hypopigmented MF is an uncommon clinical variant of MF characterized by hypopigmented macules and papules without atrophy (Fig. 2A). It is more common in patients with a dark complexion and is one of the most frequent variants seen in childhood, although cases have also been reported in adults.13 Hypopigmented MF has a better prognosis than the classic form of the disease and it responds favorably to phototherapy with narrowband UV-B, particularly in juvenile-onset MF.14 Lesions gradually regain pigmentation following treatment. Although hypopigmented lesions are sometimes the only manifestation of MF, a thorough physical examination often reveals the presence of characteristic erythematous patches or plaques.

Hypopigmented mycosis fungoides. A, Photographs showing hypopigmented macules and plaques without cutaneous atrophy in a child. B, Panoramic view showing an infiltrate in the superficial dermis. C,D, Detailed view of the dermal infiltrate with atypical lymphocytes and epidermotropism.
Classic histopathologic features of MF are common in the hypopigmented variant, which is characterized by marked epidermotropism frequently associated with atypical CD8+ T cells. Occasional nonspecific findings include a periadnexal and perivascular dermal lymphocytic infiltrate and psoriasiform hyperplasia in the epidermis (Fig. 2B-D). The characteristic immunophenotypic profile of hypopigmented MF is decreased CD7 expression and a greater presence of CD1a+ Langerhans cells in the epidermis, although CD4+ cells have also been described. CD30 expression is rare.13,15
The main entity that should be contemplated in the differential diagnosis is inflammatory vitiligo,16,17 which is also characterized by an epidermotropic CD8+ T-cell infiltrate. Integration of clinical and pathologic findings and regular follow-up are often necessary to distinguish between hypopigmented MF and inflammatory vitiligo. It is important to include other skin disorders with hypopigmentation (e.g., pityriasis alba) in the differential diagnosis.
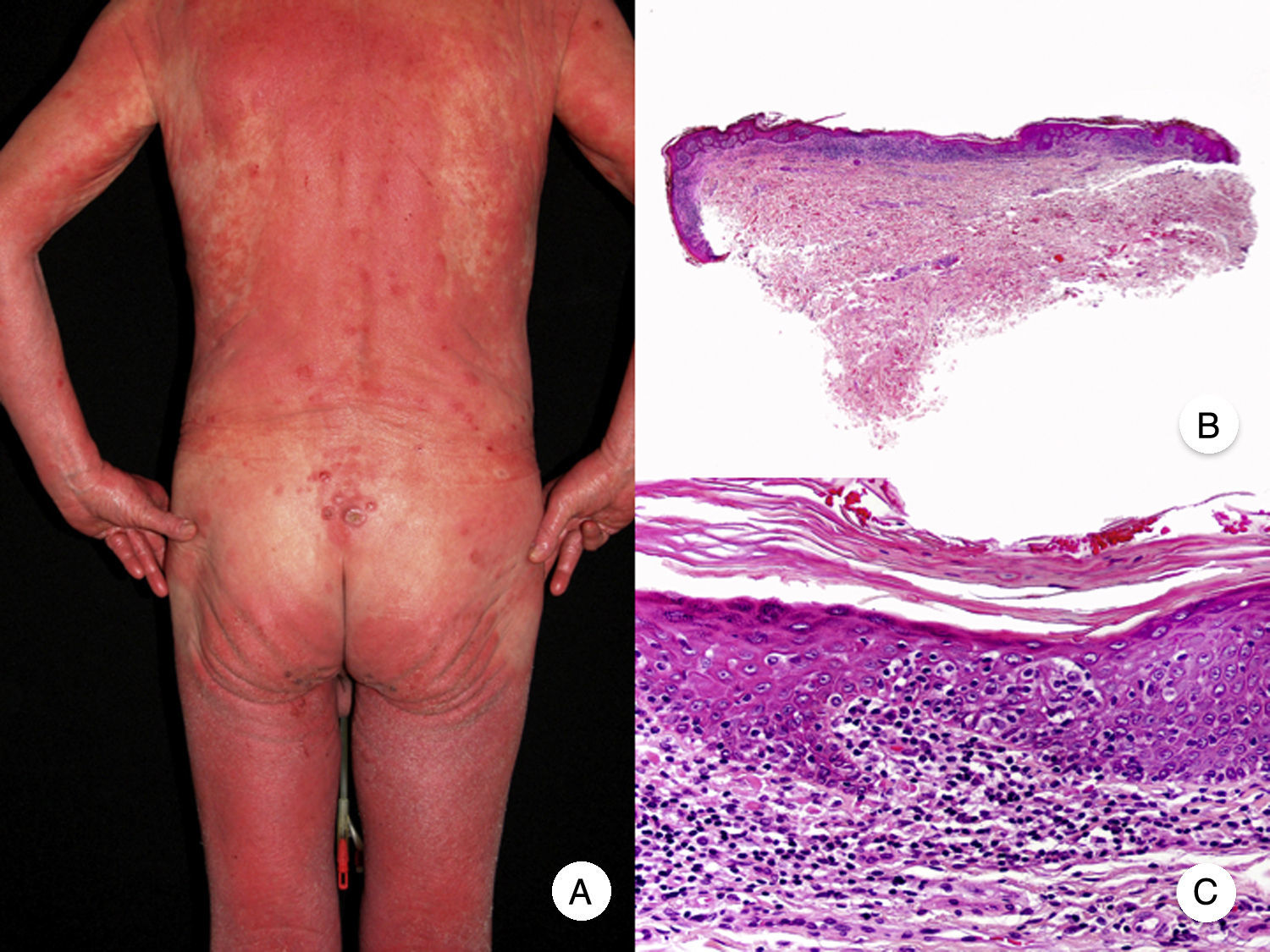
Erythrodermic MFErythrodermic MF is a variant of MF in which patients with classic histopathologic features of MF develop erythroderma but do not meet the diagnostic criteria of Sézary syndrome (Fig. 3).18 Although on rare occasions it may be the first manifestation of MF, erythroderma typically appears after the development of the characteristic patches and plaques. It is frequently associated with inappropriate treatments, facilitating diagnosis.19 Classic MF lesions reappear in patients with erythrodermic MF following appropriate treatment of erythroderma.

Erythrodermic MF can be distinguished from Sézary syndrome, as patients will not have the typical blood alterations seen in Sézary syndrome; they may have enlarged lymph nodes but this is uncommon. Although both entities share hematology-oncology staging and classification criteria,20,21 recent studies have shown that they have different chromosomal alterations22 and immunophenotypic profiles,23 demonstrating that they originate from 2 distinct populations of lymphocytes and thus constitute separate entities.
Ichthyosiform MFIchthyosiform MF is an uncommon clinical variant of MF. It typically affects the extremities and is characterized by geometric plaques with a cobblestone pattern similar to that seen in ichthyosis vulgaris.24 Ichthyosiform MF occasionally occurs in association with follicular papules and other characteristic lesions of folliculotropic MF.
Ichthyosiform MF has similar histopathologic findings to classic MF combined with features of ichthyosis, such as hypogranulosis and hyperkeratosis.25 The detection of features from both entities in the same biopsy specimen narrows the differential diagnosis down to ichthyosiform MF and paraneoplastic ichthyosis in a patient with known MF. Histopathologic examination of paraneoplastic ichthyosis lesions will only show findings associated with ichthyosis, i.e., the characteristic lymphocytic infiltrate seen in MF will be absent. Infiltration of hair follicles by atypical lymphocytes, associated or not with follicular papules, may sometimes be seen, suggesting perhaps a closer association with folliculotropic MF.25
MF Palmaris et PlantarisMF palmaris et plantaris, another uncommon clinical variant of MF, is characterized by the exclusive involvement of the palms and soles in the form of patches, plaques, or tumors.26 The lesions can spread to the dorsum of the hands and feet and to the fingers, wrists, and toes. Onychodystrophy is common. A patient with MF palmaris et plantaris will have characteristic MF lesions in acral regions only. The condition follows an indolent course and involvement of deeper areas of the skin is unusual.27
The histologic findings of MF palmaris et plantaris are again similar to those seen in classic MF, contrasting with the atypical pagetoid lymphocytes and striking epidermotropism seen in pagetoid reticulosis (Woringer-Kolopp disease).
Papillomatous MFFilamentous or vegetating MF lesions are reminiscent of acanthosis nigricans or seborrheic keratosis; they are generally located in flexural regions such as the neck, the axillae, and the inguinal folds. Histopathologic findings include marked acanthosis and papillomatosis together with a band-like infiltrate composed of atypical lymphocytes that may or may not be epidermotropic.28,29
Papular MFPapular MF is a clinical variant of MF characterized by the presence of small non-folliculocentric papules and the absence of conventional MF patches and plaques (Fig. 4A).30 Histopathologic findings are similar to those observed in classic MF and have a characteristic patch-like distribution (Fig. 4 B-D); there is no follicular involvement. Although papular MF was originally described as a benign condition with a favorable long-term prognosis,31 there have been reports of erythroderma appearing after relatively short periods of time and of rapid progression to the tumor stage.32

Diagnosis is challenging given the absence of the typical clinical manifestations of MF. The main entities to consider in the differential diagnosis are lymphomatoid papulosis and other primary cutaneous lymphoproliferative disorders. Papular MF lesions tend to be stable and characteristic features seen in lymphomatoid papulosis (spontaneous regression, ulceration, hemorrhaging, and residual varioliform scars) are absent. The infiltrate in papular MF typically has a CD30− immunophenotype.33
Solitary or Unilesional MFSolitary or unilesional MF refers to an isolated macule, plaque, or nodule with indistinguishable histopathologic characteristics to those of classic MF; there is no evidence of any other cutaneous lesions.34,35 The observation of a band-like inflammatory infiltrate accompanied by isolated epidermotropic atypical lymphocytes but no other histopathologic features requires consideration of localized pagetoid reticulosis, another variant of MF that also manifests as isolated lesions but has different clinical and histologic features.36 Solitary or unilesional MF is associated with a favorable prognosis; it follows a benign course and rarely progresses to more advanced forms.
Invisible MFInvisible MF is a variant of MF in which histopathologic features of classic MF are detected in normal-appearing skin; there are no visible skin lesions and the only symptom is pruritus. Skin lesions may remain absent for the entire course of the disease.37
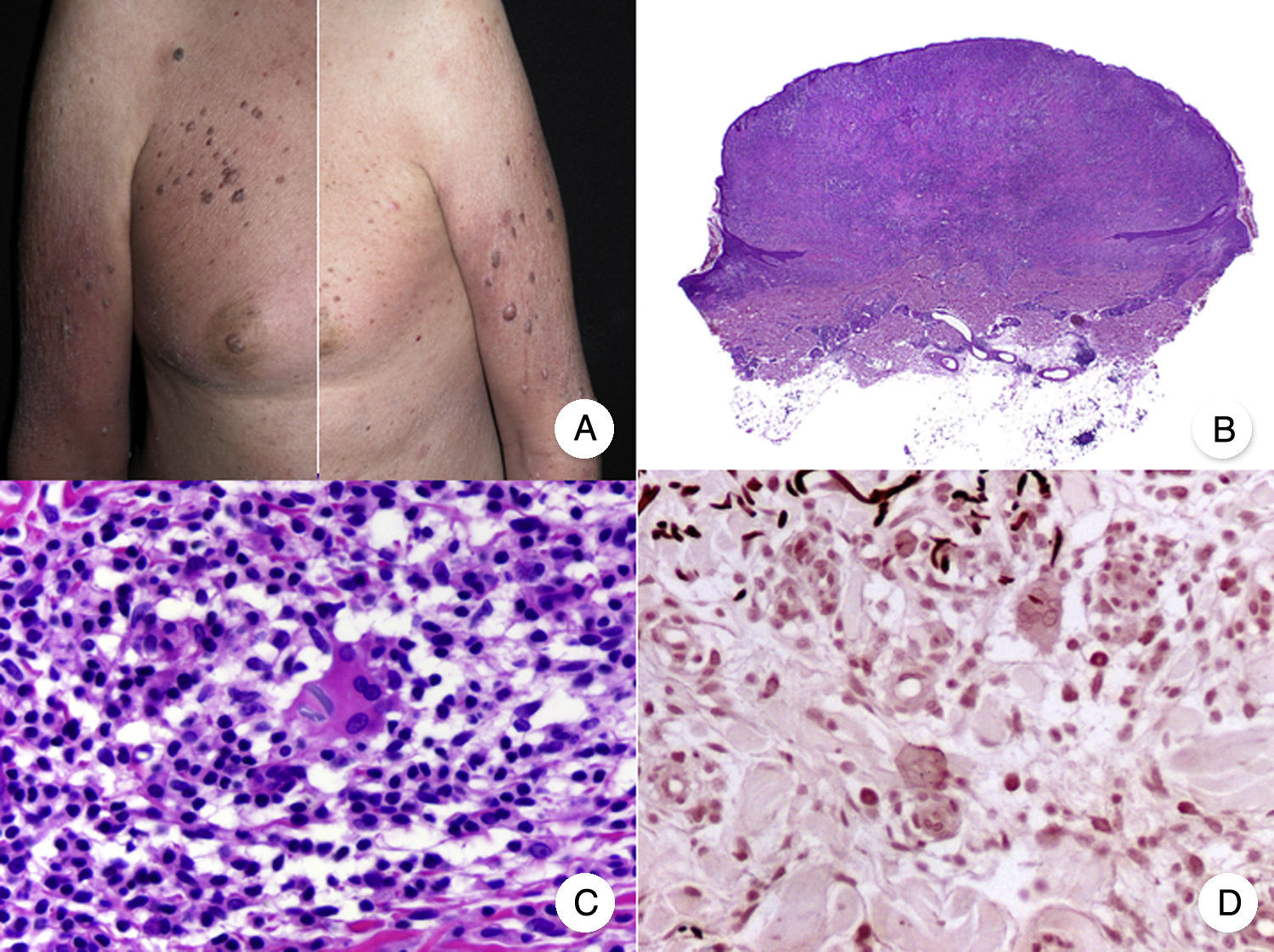
Clinicopathologic Variants of MFFolliculotropic MFFolliculotropic MF is one of the most common variants of MF and has accounted for up to 10% of all forms of MF in some series.38 It is considered to be an entity in its own right, and was included in the WHO-EORTC (European Organization for Research and Treatment of Cancer) classification for cutaneous lymphomas20 and in the 2016 revision of the WHO classification of hematopoietic and lymphoid tumors.21
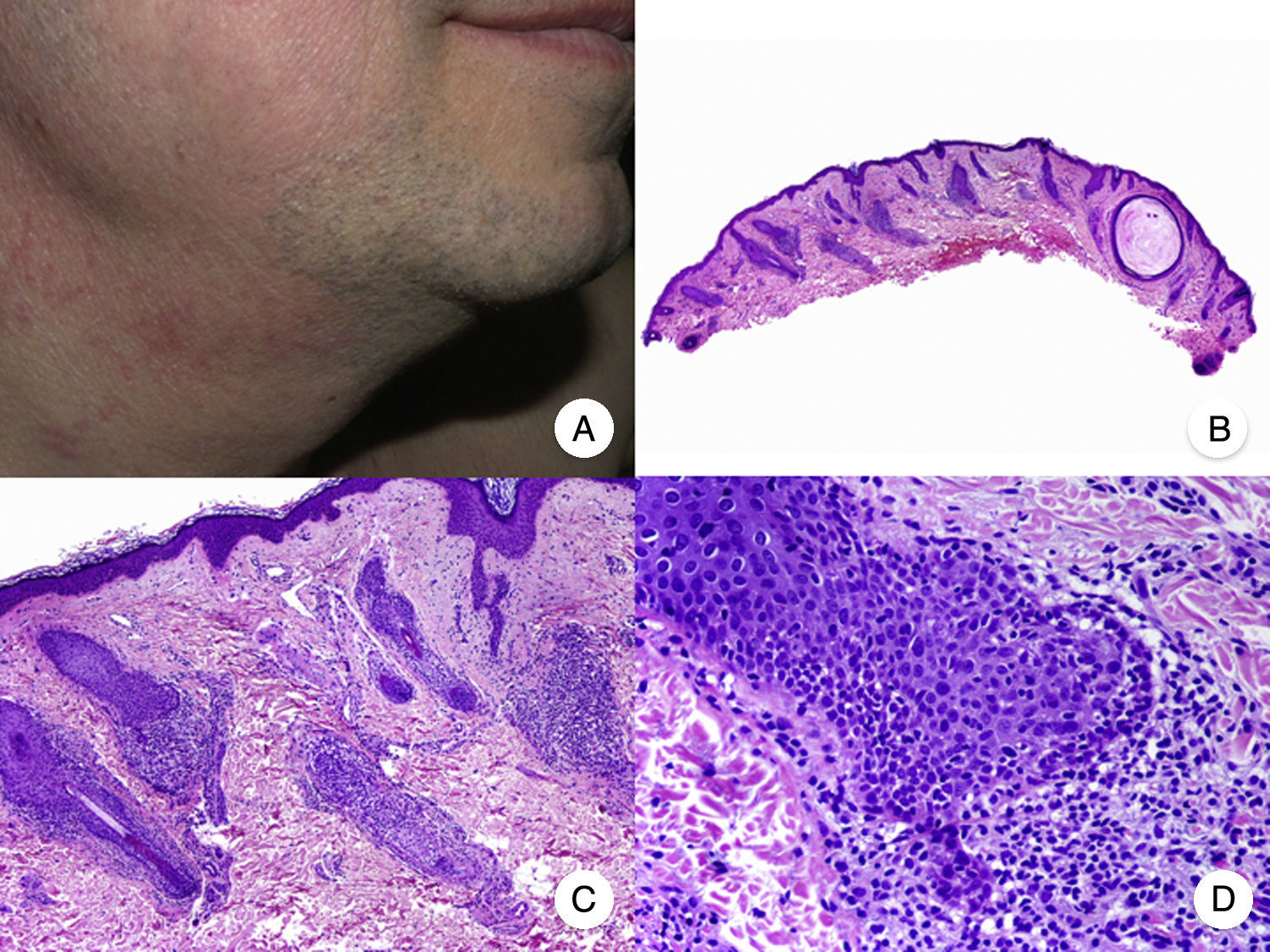
Typical clinical manifestations of folliculotropic MF are indurated erythematous plaques combined with follicular papules and acneiform lesions including small cysts and comedones. Lesions are typically located on the head and neck and are frequently associated with alopecia and on occasions with mucinorrhea (Fig. 5A). Infiltrated plaques in the eyebrows with hair loss are also common. Folliculotropic MF is usually highly pruritic. The pruritus tends to be refractory to treatment and, as occurs in tumor-stage MF, is associated with poor prognosis.39,40 The folliculotropic variant of MF mainly affects adults (predominantly males), although cases have been described in children and adolescents.20
. A, Photograph showing follicular papules and acneiform lesions on the face and neck. B, Panoramic view showing an infiltrate around the hair follicles. C, D, Higher-magnification view showing aggregates of atypical lymphocytes in the outer root sheath of the hair follicle.")
Folliculotropic mycosis fungoides (MF). A, Photograph showing follicular papules and acneiform lesions on the face and neck. B, Panoramic view showing an infiltrate around the hair follicles. C, D, Higher-magnification view showing aggregates of atypical lymphocytes in the outer root sheath of the hair follicle.
Histopathologic features of folliculotropic MF include isolated or aggregates of atypical lymphocytes in the outer root sheath of the hair follicle (a phenomenon known as folliculotropism), in addition to a perivascular and periadnexal dermal inflammatory infiltrate of atypical lymphocytes, eosinophils, and plasma cells (Fig. 5 B-D). Although isolated lymphocytes may be seen dotted through the epidermis, epidermotropism is not common in folliculotropic MF.41 A significant inflammatory infiltrate involving the eccrine sweat glands may be observed and should raise suspicion of extensive adnexal involvement. Another common finding is mucinous degeneration of the follicular epithelium (follicular mucinosis), which becomes more evident on staining with Alcian blue or other mesenchymal mucin stains. Immunohistochemical findings are similar to those seen in classic MF and CD30+ infiltrates are common.20
MF With Eruptive Infundibular CystsA localized or generalized follicular eruption consisting of infundibular cysts and comedones has been described in some patients with MF and appears to be a rare manifestation of the disease.42–44 Because of their size and inflammatory appearance, the lesions may sometimes simulate tumor-stage lesions.45 MF with eruptive infundibular cysts must be distinguished from folliculotropic MF, although some authors consider the 2 variants to be part of the same spectrum of follicular involvement in MF.32
Histology shows the characteristic features of an infundibular cyst surrounded by a dense infiltrate formed primarily by atypical lymphocytes in the cyst wall. The appearance of infundibular cysts may be related to infiltration of follicular openings by neoplastic cells, causing subsequent blockage and dilation of the infundibula.42
Syringotropic MFSyringotropic MF is an uncommon clinicopathologic variant of MF characterized by eccrine gland involvement. Clinical manifestations include erythematous papules and plaques that may or may not be accompanied by a follicular eruption. The papules and plaques are sometimes hyperpigmented. Adnexal involvement frequently leads to anhidrosis and alopecia.46 Patients can have solitary or multiple syringotropic lesions, which may be accompanied by classic MF lesions in other areas of the skin. Palmoplantar involvement is common. Syringotropic MF follows an indolent clinical course.
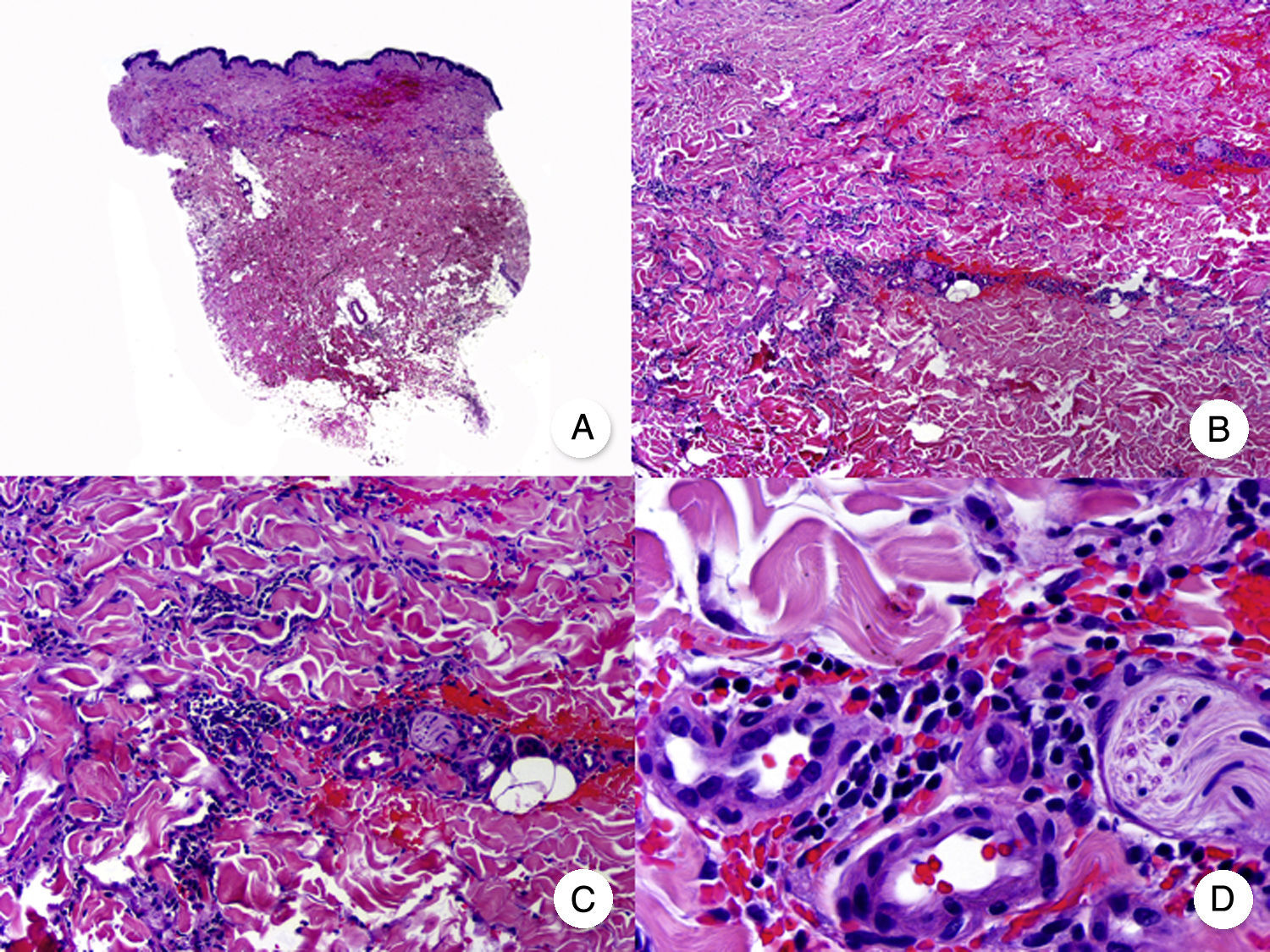
The most characteristic histologic findings, which allow the condition to be distinguished from folliculotropic MF, are a dense infiltrate of atypical lymphocytes in the eccrine glands and ducts and a variable degree of syringometaplasia (squamous transformation of the glandular epithelium) (Fig. 6).47 Epidermotropism and follicular involvement are common.46 Other less specific findings are a band-like inflammatory infiltrate and epidermal hyperplasia. The condition traditionally known as syringolymphoid hyperplasia is now considered to be syringotropic MF.

Syringotropic mycosis fungoides. A, Panoramic view showing band-like infiltrate in the reticular and periglandular dermis. B, Higher-magnification view showing dense infiltration of eccrine glands by lymphocytes. C, D, Higher-magnification view showing atypical lymphocytes surrounding an eccrine sweat gland with syringometaplasia.
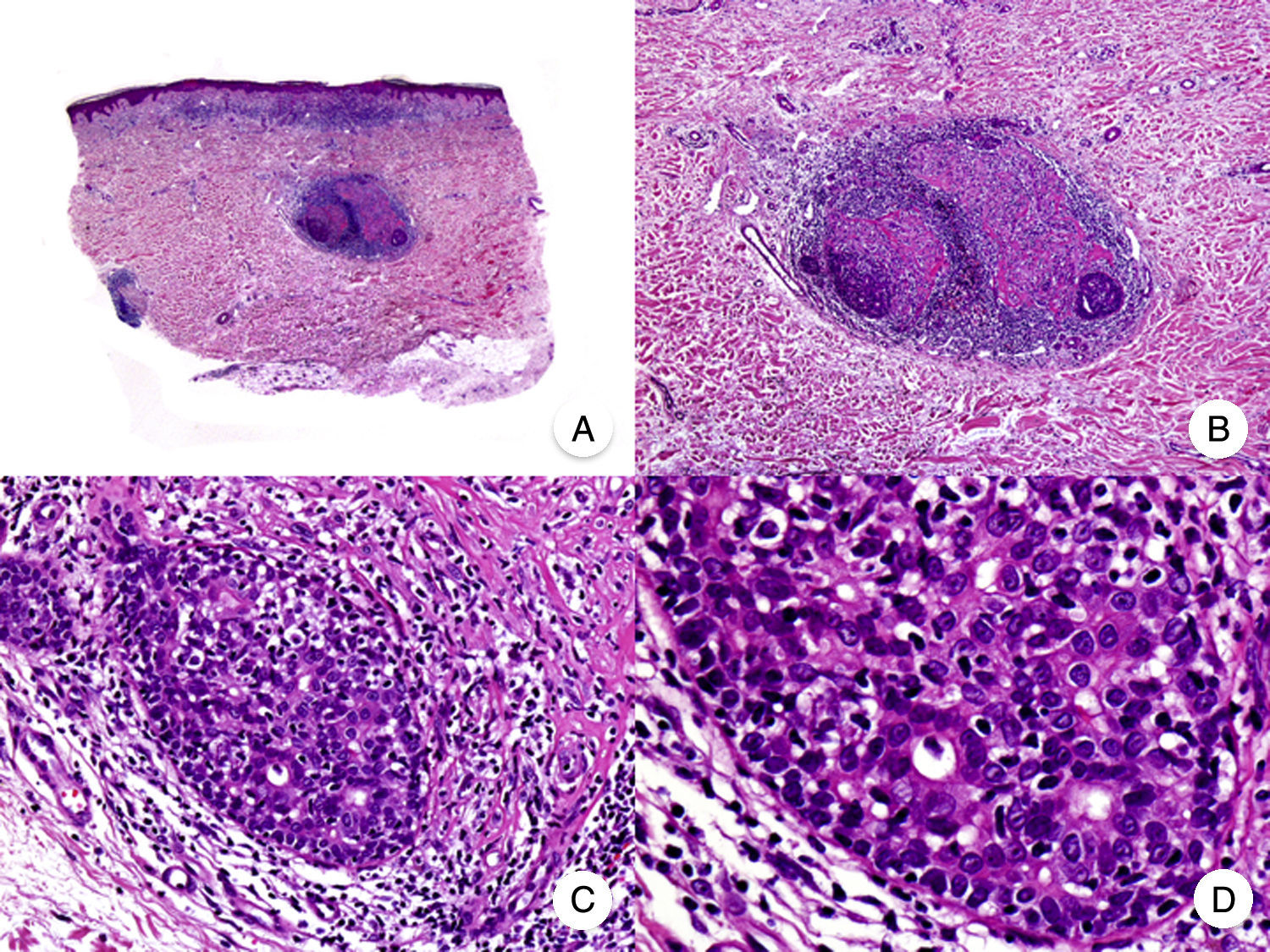
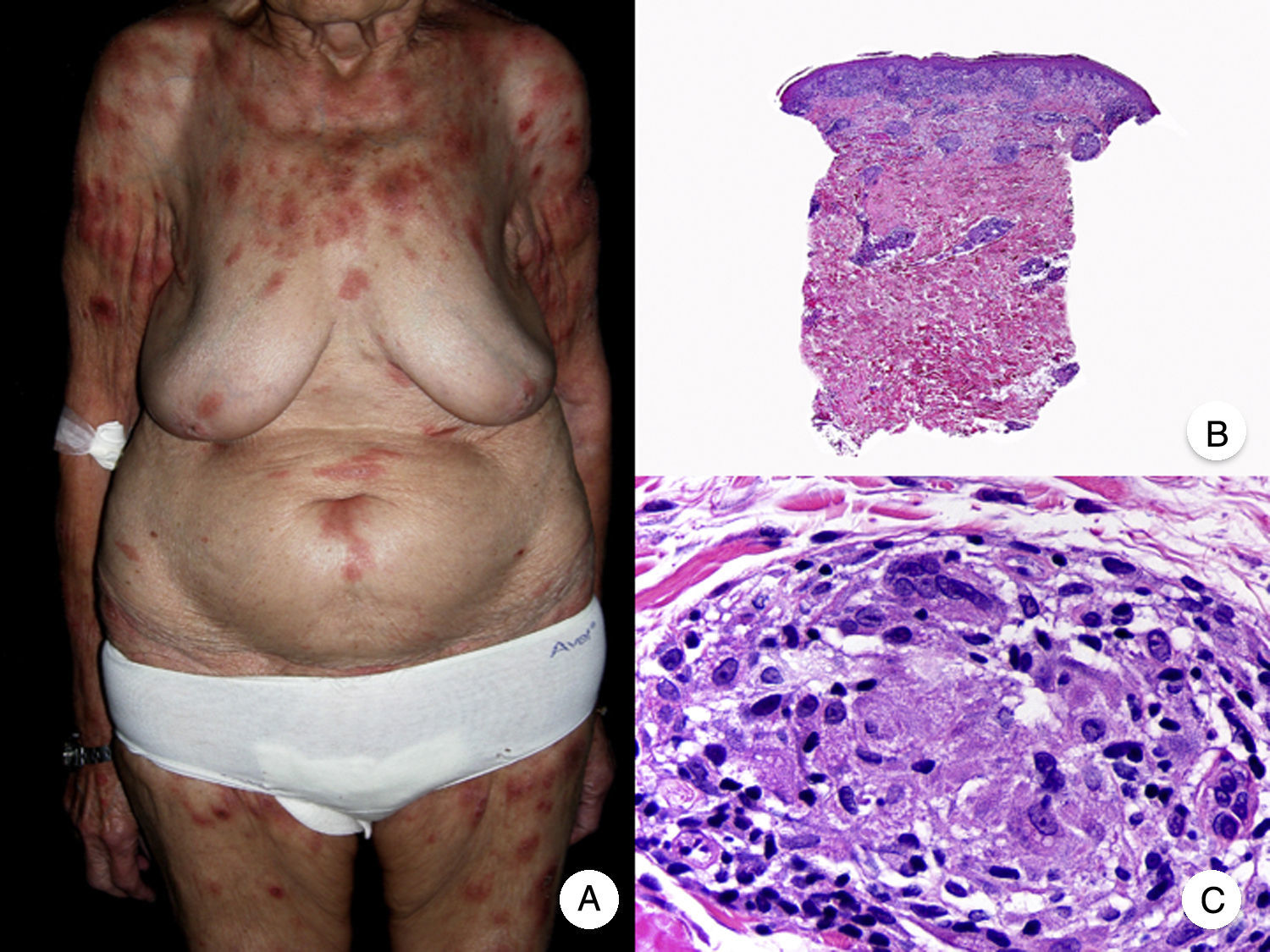
Granulomatous slack skin is a very rare variant of MF. It is characterized by erythematous plaques that take the form of frequently large folds of lax pendulous skin generally located in flexural areas such as the axillae and groin (Fig. 7A).48 Its distinctive clinical and histopathologic features earned it a place in the latest WHO-EORTC classification for cutaneous lymphomas.20,21 It follows an indolent course and tends to recur after local excision. It is more common in young patients. Many patients with granulomatous slack skin have a second lymphoma, and Hodgkin disease is particularly common.20

Granulomatous slack skin. A, Photograph showing an erythematous plaque in the form of a large pendulous skinfold in the axilla. B, Panoramic view showing a diffuse inflammatory infiltrate throughout the dermis and extending into the subcutaneous tissue. C, Higher-magnification view showing that the infiltrate is composed of histiocytes, atypical lymphocytes, and giant multinucleated cells with many nuclei.
Characteristic histopathologic findings include a diffuse granulomatous inflammatory infiltrate that occupies the full thickness of the dermis and occasionally extends into the subcutaneous tissue. The infiltrate is composed of histiocytes, atypical lymphocytes, and giant multinucleated cells, which often contain large numbers of nuclei. Another characteristic finding is a striking loss, or even total absence, of elastic fibers, fragments of which can be observed in the interior of the giant multinucleated cells (elastophagocytosis) (Fig. 7B and C). Giant multinucleated cells also frequently contain lymphocytes as a result of phagocytosis or emperipolesis and there is a notable absence of epidermotropism and Pautrier microabscesses. Granulomatous MF is distinguished by the presence of small sarcoid granulomas dotted through the dermis and the absence of elastophagocytosis. However, the 2 variants are essentially distinguished on clinical grounds.49
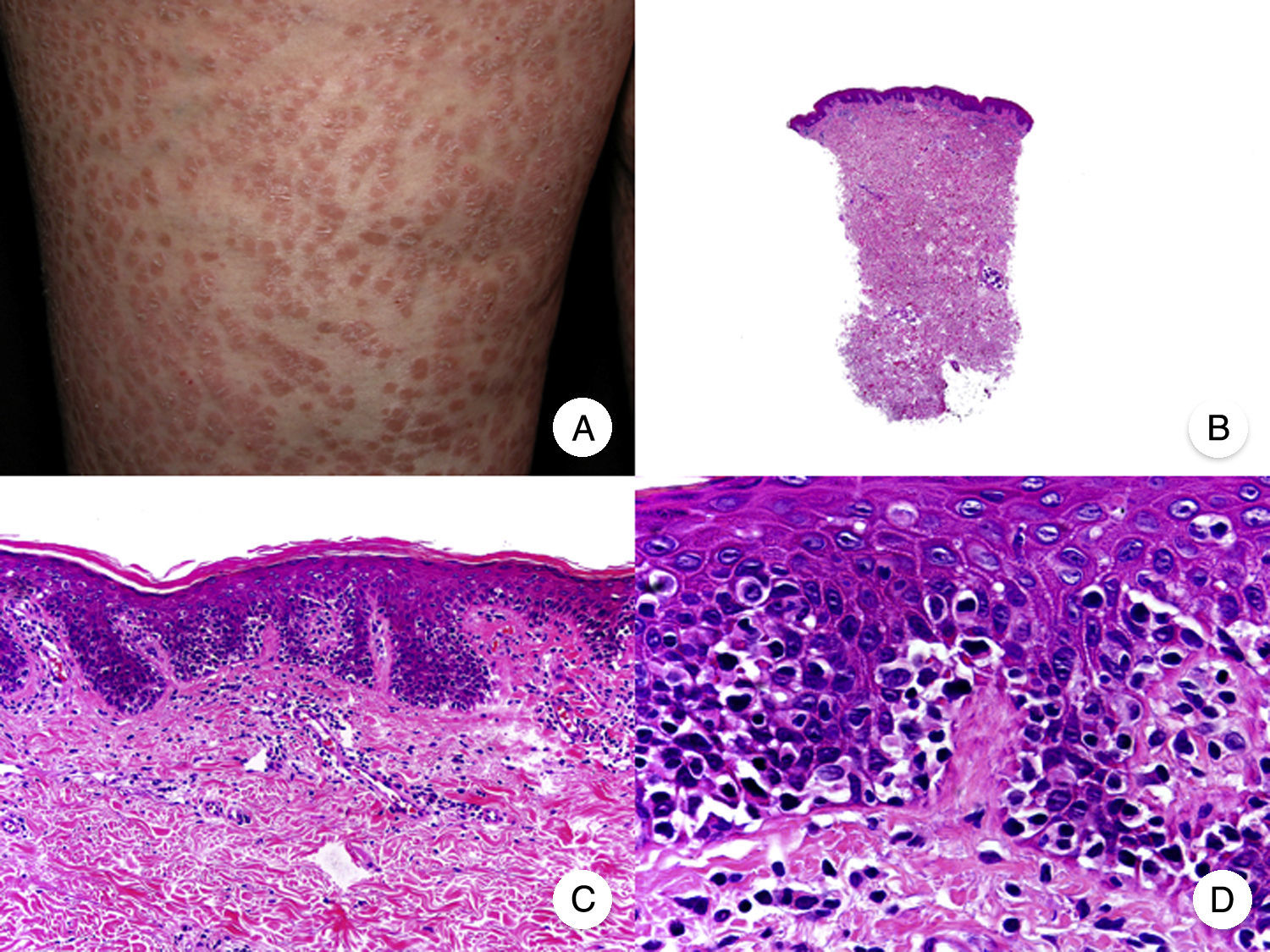
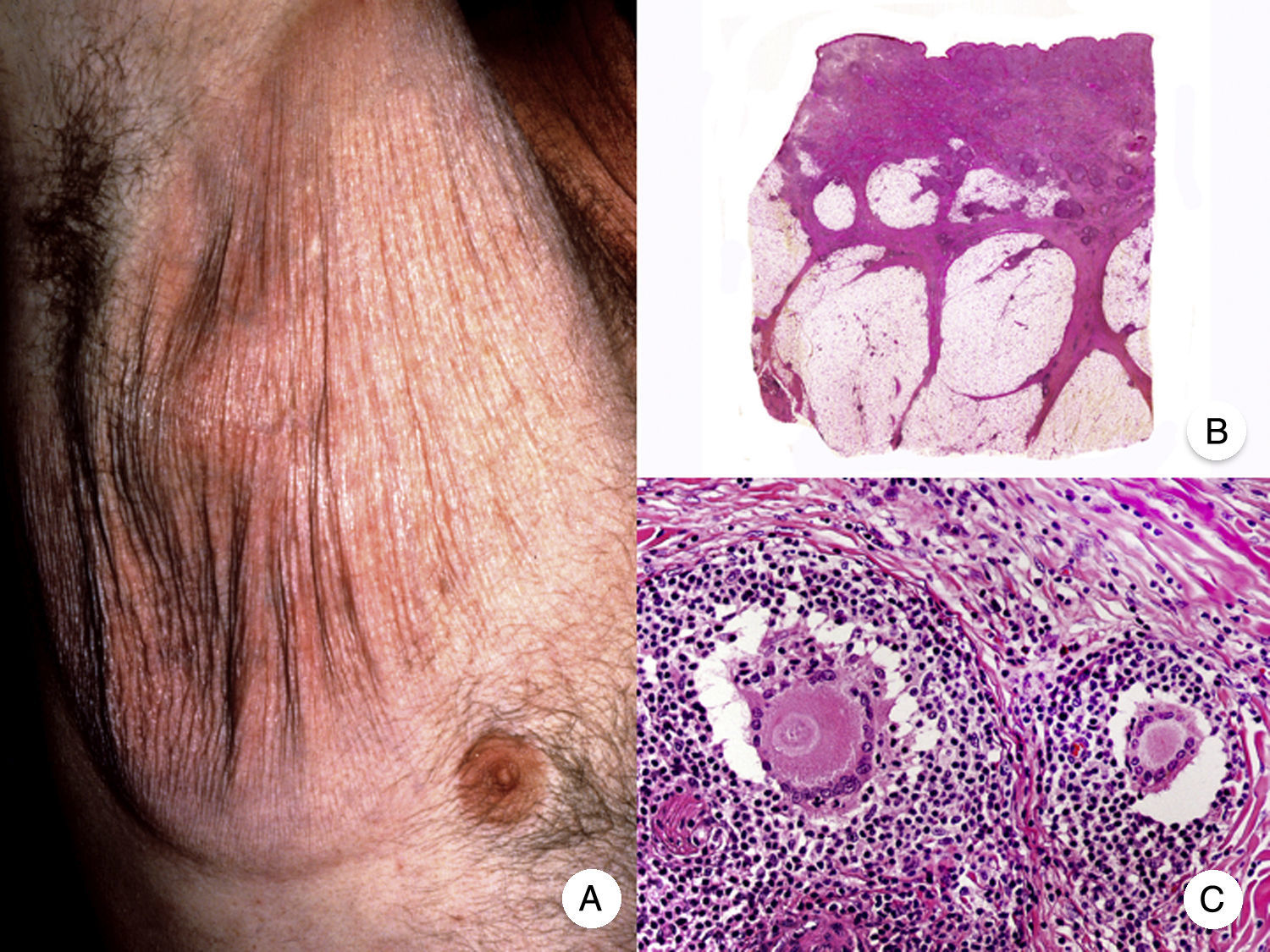
Pagetoid ReticulosisPagetoid reticulosis or Woringer-Kolopp disease is a rare variant of MF characterized by slow growth, an indolent clinical course, and a favorable prognosis. It is included in the latest WHO-EORTC classification for cutaneous lymphomas.20,21 It clinically manifests as a solitary lesion generally located in the acral areas of the extremities. The lesion is typically an erythematous plaque with a psoriasiform, hyperkeratotic appearance (Fig. 8A). The treatment of choice is surgical excision or local radiation therapy. The presence of disease progression or extracutaneous involvement, classically referred to as disseminated pagetoid reticulosis or Ketron-Goodman disease, is currently considered to correspond to a more aggressive cutaneous lymphoma (epidermotropic cytotoxic T-cell lymphoma in nearly all cases).20

Pagetoid reticulosis. A, Photograph showing a solitary erythematous, hyperkeratotic, psoriasiform plaque on the heel. B, Panoramic view showing epidermal hyperplasia, hyperkeratosis, and parakeratosis and an infiltrate in the papillary dermis. C,D, Higher-magnification view showing the marked epidermotropism of the infiltrate, formed by atypical pagetoid lymphocytes with large hyperchromatic nuclei.
Characteristic histopathologic findings of pagetoid reticulosis include an infiltrate with marked epidermotropism composed of atypical pagetoid lymphocytes with a large hyperchromatic nucleus surrounded by a pale halo, in addition to epidermal hyperplasia with hyperkeratosis and parakeratosis (Fig. 8 B-D). Pagetoid reticulosis is often characterized by a CD8+ immunophenotype with CD30 expression in most cases,50 although this is not associated with a more aggressive biologic behavior.51
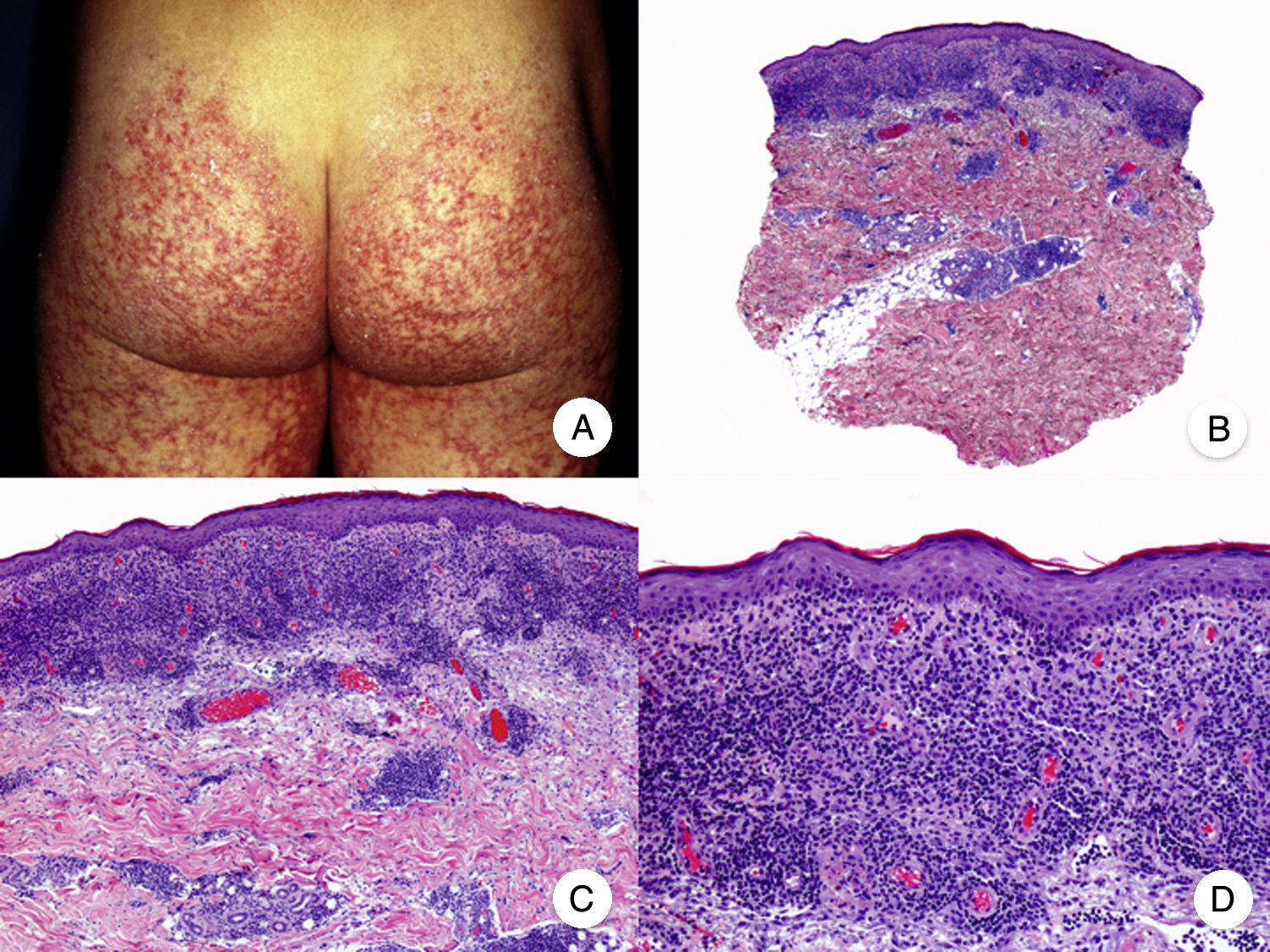
Poikilodermal MFPoikilodermal MF, which is classically referred to as poikiloderma vasculare atrophicans, is one of the most common variants of MF. It manifests as plaques with atrophy, hyperpigmentation, hypopigmentation, and telangiectasia covering a large area of skin. Compared with other variants of MF, prognosis appears to be favorable and the lesions respond well to phototherapy (Fig. 9A).52 The most common areas affected are the breasts in women and the buttock region in women and men.

Poikilodermal mycosis fungoides. A, Photograph showing plaques with atrophy, hyperpigmentation, hypopigmentation, and telangiectasia on both buttocks. B, Panoramic view showing a lichenoid infiltrate in the papillary dermis. C, D, Higher-magnification view showing epidermal atrophy with flattening of the dermal-epidermal junction, an infiltrate composed of atypical lymphocytes with epidermotropism and dilatations, and dilated capillaries in the superficial dermis.
Histopathologic features of poikilodermal MF include epidermal atrophy with flattening of the dermal-epidermal junction, vacuolar degeneration of the basal layer, and a lichenoid epidermotropic infiltrate composed of atypical lymphocytes. Other relevant findings are apoptotic keratinocytes, pigmentary incontinence, and dilated capillaries in the superficial dermis (Fig. 9 B-D). Pautrier microabscesses are not usually present. Neoplastic cells frequently express a CD8+ immunophenotype.52
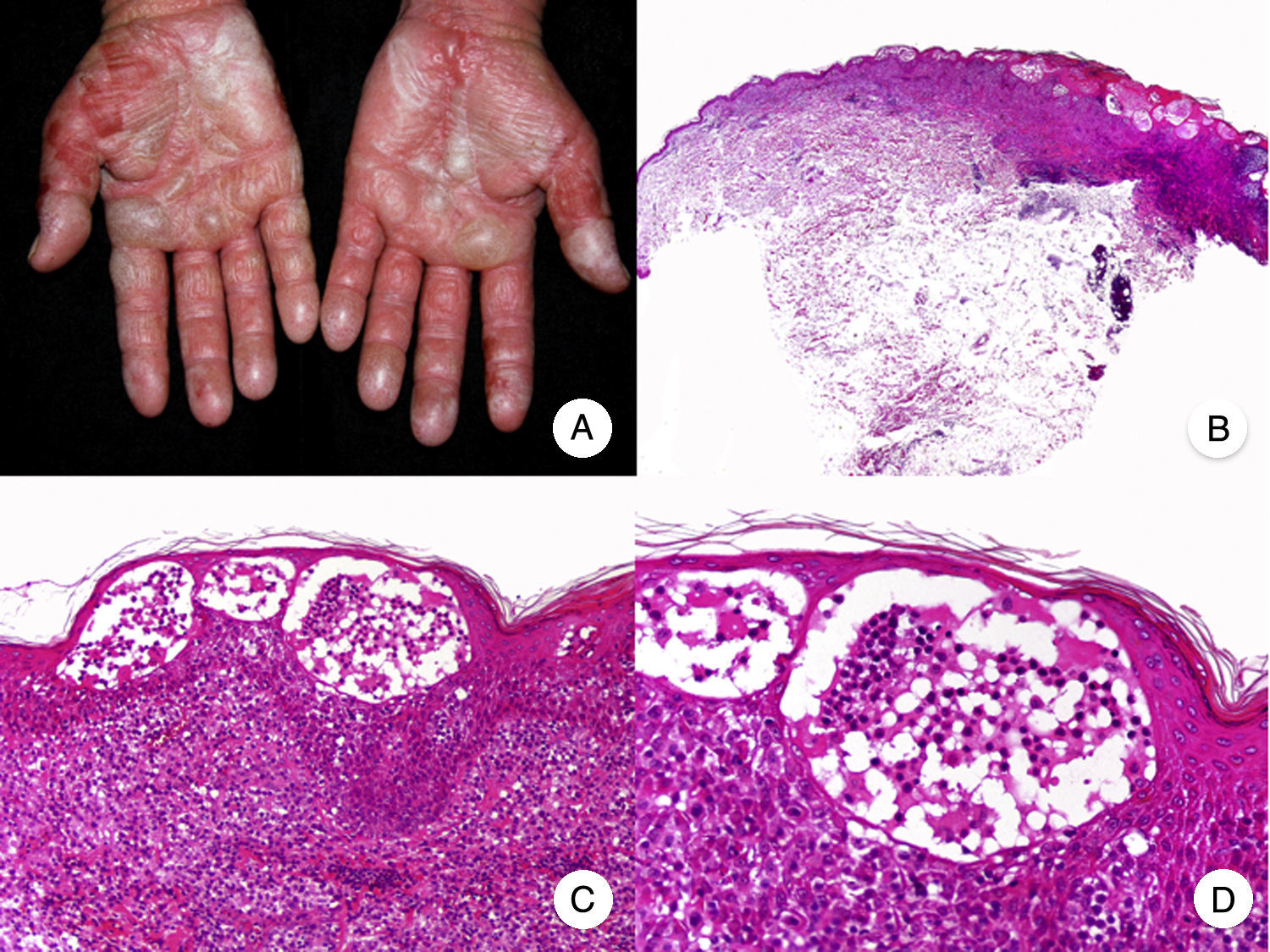
Bullous MFBullous MF is a clinicopathologic variant of MF characterized by bullous vesicular lesions that was described by Kaposi in 1887.53 The lesions can be flaccid or tense and they usually affect large areas of the trunk and extremities (Fig. 10A). The rupture of bullae can cause superficial erosions. Bullous lesions frequently coexist with classic MF lesions and can appear either as the first manifestation of MF or later.53,54 The term dyshidrotic MF has been used to describe bullae confined to the palms and soles.55

Histopathologically, bullous MF is characterized by intraepidermal or subepidermal blisters combined with classic features of MF, such as atypical lymphocytes, epidermotropism, and Pautrier microabscesses (Fig. 10 B-D). Direct and indirect immunofluorescence results are negative, helping to distinguish this variant from autoimmune blistering diseases. Other causes of bullous lesions, such as drugs or bacterial or viral infections, must be contemplated in the differential diagnosis.53,56
Prognosis is poor and the 1-year survival rate following onset of bullous-vesicular lesions is approximately 50%.53,57
Anetodermic MFThe term anetoderma refers to the progressive loss of dermal elastic tissue that results in atrophic plaques with a characteristic parchment-like surface (Fig. 11A). Anetoderma arising in classic MF lesions is very rare and has only been described in 2 patients to date.58

A, Photograph showing a patient with atrophic plaques and atrophic nodules with a parchment-like surface. B, Panoramic view of a diffuse infiltrate occupying the entire dermis. C, Detailed view at a higher magnification of the infiltrate, showing atypical lymphocytes and elastophagocytosis. D, Orcein stain showing the absence of elastic fibers in the dermis.
The histopathologic findings observed in the 2 patients were similar to those seen in classic MF, but elastic tissue stains (e.g., orcein and Van Gieson) also showed absence of elastic fibers in the dermis (Fig. 11 B-D). Contrasting with granulomatous slack skin, elastophagocytosis is an uncommon focal finding in anetodermic MF.58
Hyperpigmented MFHyperpigmented MF is a very uncommon variant of MF in which hyperpigmented macules and plaques are often the only clinical manifestation of disease. Hyperpigmented MF is occasionally seen in association with other uncommon variants of MF, but in this case, the hyperpigmentation is not due to poikilodermic changes or residual hyperpigmentation from previous lesions.59
Histopathologic features include pigmentary incontinence with abundant melanin granules in keratinocytes and Langerhans cells60 together with numerous melanophages in the papillary dermis and the classic features of MF. Neoplastic cells tend to be located around the dermal-epidermal junction and display a characteristic CD8+ immunophenotype.61
Hyperpigmented MF appears to follow an indolent, relatively nonaggressive course.59,61
Purpuric MFPurpuric MF is clinically characterized by persistent purpuric lesions and histologically characterized by a band-like infiltrate composed mainly of atypical lymphocytes together with hemosiderin-containing macrophages and extravasated erythrocytes. The purpuric lesions progress to the typical lesions seen in MF and this variant is more common in males.62,63
The greatest diagnostic challenge lies in distinguishing purpuric MF from other benign purpuric skin disorders as clinical and on occasions histopathologic findings overlap; a putative relationship between chronic pigmented purpuric dermatosis and MF has been proposed.63–65
Pustular MFThe term pustular MF refers to an extremely rare clinicopathologic variant of MF that was initially described by Ackerman66 as a long-standing vesicular pustular eruption that progressively gives way to typical MF plaques. The pustules can be generalized or confined to the palmoplantar surfaces.67,68
Histopathologic examination shows typical MF features, such as a band-like infiltrate of atypical lymphocytes, epidermotropism, and Pautrier microabscesses, in association with subcorneal pustules containing atypical lymphocytes, neutrophils, and eosinophils.68 The proportion of neoplastic and inflammatory cells is variable, but the latter predominate.66
Verrucous MFVerrucous or hyperkeratotic MF, another rare variant of MF, is characterized by the presence of localized or disseminated verrucous hyperkeratotic lesions that affect acral surfaces and may coexist with typical MF lesions.24,69,70 The differential diagnosis should include palmoplantar MF confined to these regions. Histopathologic findings include classic MF features in addition to a perivascular inflammatory infiltrate in the papillary dermis, spongiosis and exocytosis in the epidermis, and papillomatosis and hyperkeratosis.71
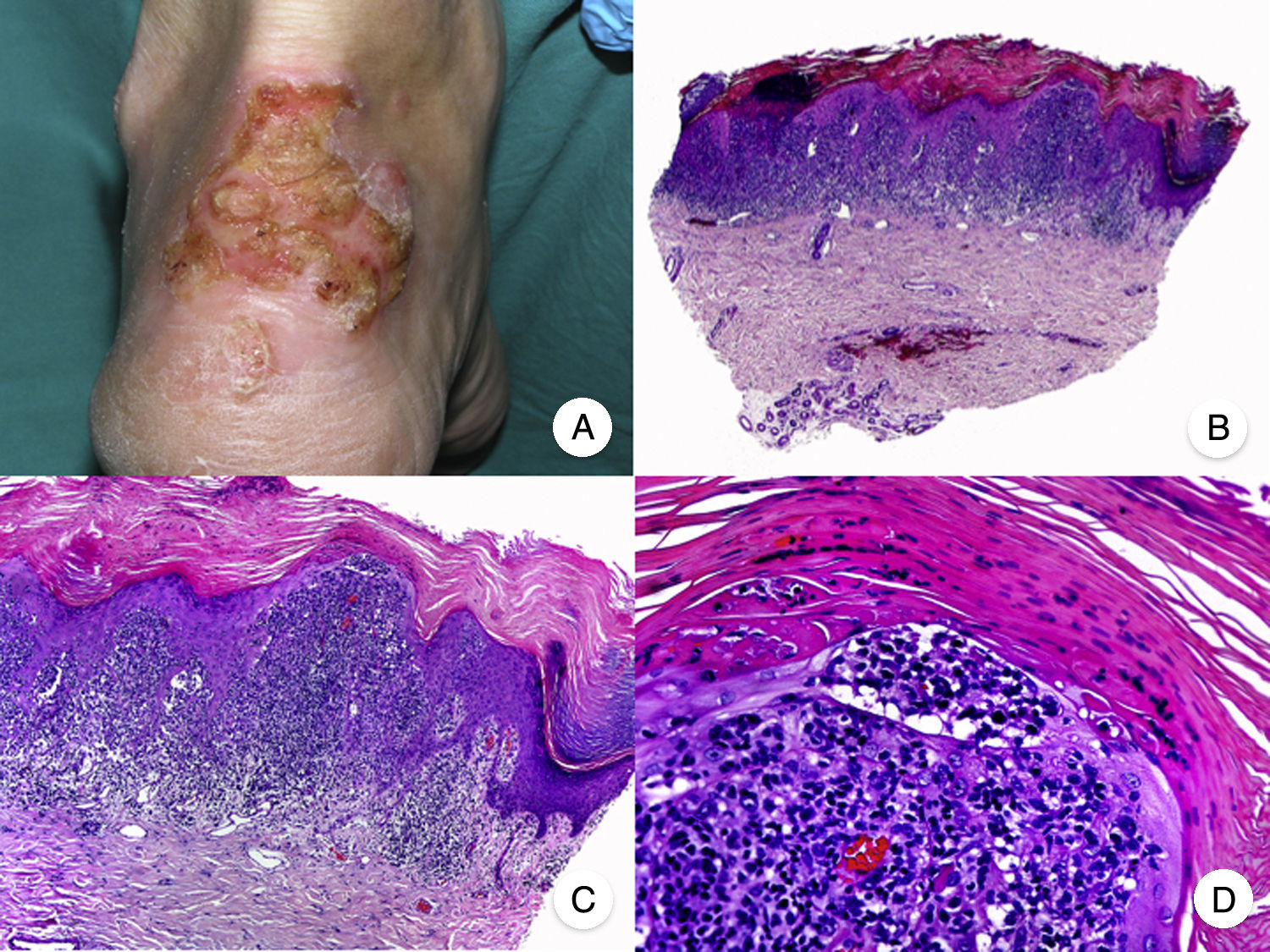
Histopathologic Variants of MFGranulomatous MFGranulomatous MF is a histopathologic variant that must be diagnosed by skin biopsy. Findings include perivascular sarcoidal granulomas throughout the dermis and an infiltrate of atypical lymphocytes, histiocytes, and giant multinucleated cells.72 Epidermotropism is observed in approximately 50% of cases, complicating diagnosis in patients without characteristic clinical manifestations.45 Some follicular involvement may be occasionally observed (Fig. 12B and C).

Granulomatous mycosis fungoides. A, Photograph of patch-stage and plaque-stage mycosis fungoides lesions distributed on the trunk and extremities. B, Panoramic view showing perivascular sarcoidal granulomas throughout the dermis. C, Detailed view of a granuloma composed of atypical lymphocytes, histiocytes, and giant multinucleated cells.
Granulomatous MF is clinically characterized by patches, plaques, and tumors similar to those seen in classic MF, but there may also be alopecia when the scalp is involved (Fig. 12A).73 Prognosis seems to be poorer than in classic MF. Granulomatous MF responds poorly to topical treatment, frequently progresses, and is associated with a high risk of a second lymphoma.73–75
Although granulomatous MF is essentially distinguished from granulomatous slack skin on clinical grounds, histopathologic examination shows a lower proportion of multinucleated cells, and elastolysis and elastophagocytosis are absent.24,49
Interstitial MFInterstitial MF is an uncommon variant of MF and it is clinically indistinguishable from classic MF. It is defined by its histopathologic features, which include a predominantly lymphocytic infiltrate and histiocytes scattered among collagen bundles in the dermis, mimicking interstitial granuloma annulare or the inflammatory phase of morphea (Fig. 13).76–78 Additional findings include epidermotropism and mucin deposits in the dermis. Pautrier microabscesses are uncommon. Interstitial MF is distinguished from granuloma annulare by its monoclonal lymphocytic population (granuloma annulare has a predominantly histiocytic infiltrate) and the observation of classic MF features in other lesions, although coexistence of MF and interstitial granuloma annulare has been described.79

Interstitial mycosis fungoides. A, Panoramic view showing a mild infiltrate in the papillary dermis. B, Higher-magnification view showing the interstitial pattern of the infiltrate. C, D, Detailed view of the predominantly lymphocytic infiltrate among the collagen bundles and other adnexal structures of the dermis.
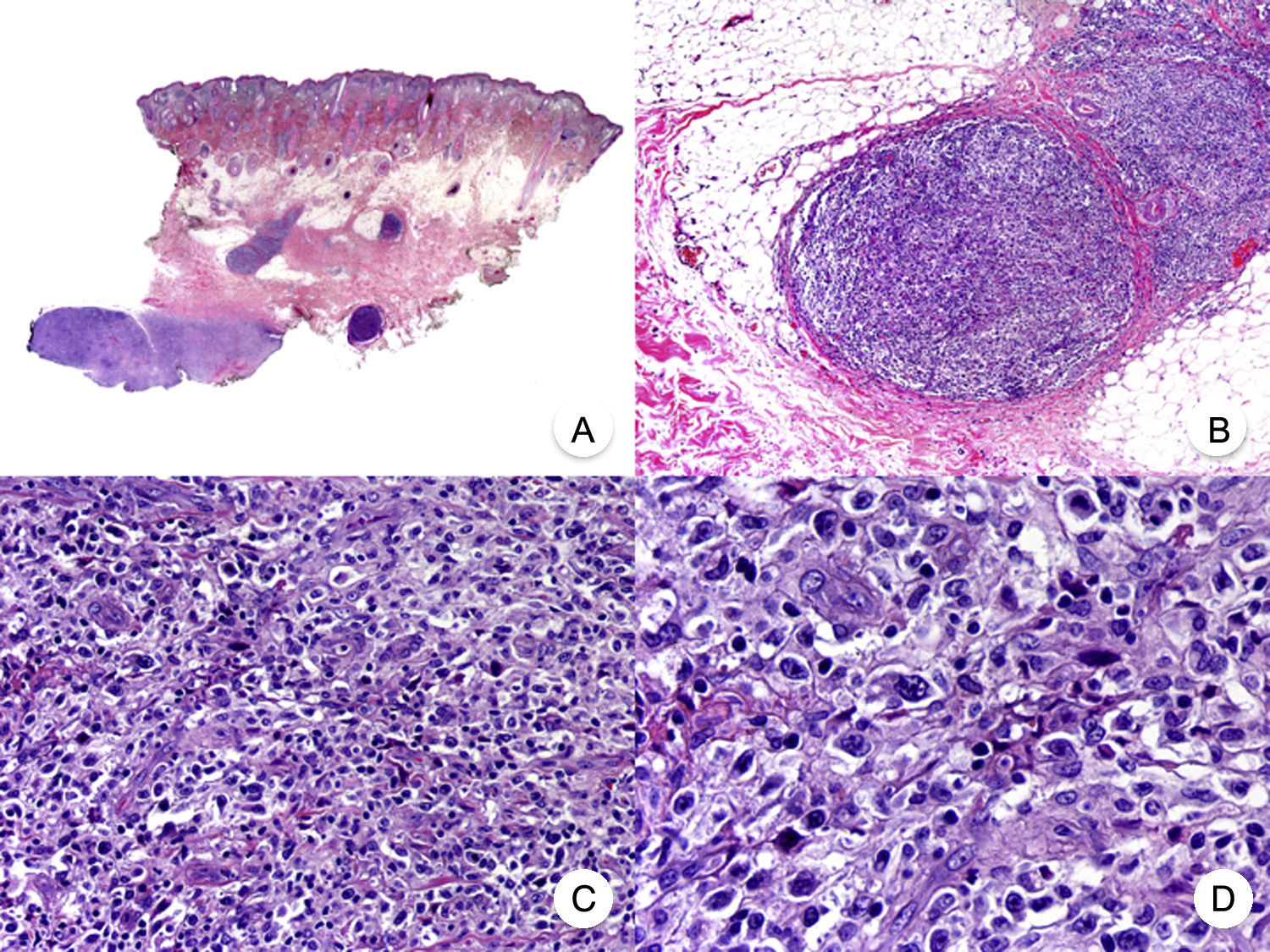
Transformation of MF into large T-cell lymphoma is a histopathologic variant of MF in which over 25% of the neoplastic infiltrate is formed by large, pleomorphic, anaplastic cells and immunoblasts; it is more common in patients with tumor-stage MF.80 CD30 expression by large cells is variable and CD30 negativity has been associated with worse prognosis (Fig. 14).81

Mycosis fungoides with large-cell transformation. A, Panoramic view of biopsy specimen from the scalp showing the patch-like infiltration of the dermis. B, Detailed view of the neoplastic infiltrate in the hypodermis. C,D, Higher-magnification view showing the large pleomorphic and anaplastic neoplastic cells in the infiltrate.
This histologic variant of MF must be distinguished from other primary cutaneous lymphomas with CD30+ large cells, such as anaplastic large-cell lymphoma and lymphomatoid papulosis. In MF, the percentage of large cells rarely reaches the 75% necessary to diagnose anaplastic large-cell lymphoma and the characteristic IRF4 translocations seen in the latter are generally not found in MF.82 The distinction with lymphomatoid papulosis must be based on clinical findings, such as the absence of spontaneous regression in MF.83
Transformation of MF into large-cell lymphoma worsens prognosis and is associated with terminal stages of the disease.81,82
Ethical DisclosuresProtection of humans and animalsThe authors declare that no tests were carried out in humans or animals for the purpose of this study.
Confidentiality of dataThe authors declare that they have followed their hospital's protocol on the publication of data concerning patients.
Right to privacy and informed consentThe authors declare that no private patient data appear in this article.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Muñoz-González H, Molina-Ruiz AM, Requena L. Variantes clínico-patológicas de micosis fungoide. Actas Dermosifiliogr. 2017;108:192–208.



