


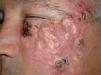

We report the case of a 66-year-old man who consulted with a 3-week history of a rapidly growing asymptomatic lesion on his left cheek that for the previous week had prevented him from opening the eye on that side (Fig. 1).

Two years earlier at another center the patient had been diagnosed with a primary systemic CD30+ anaplastic large-cell lymphoma (ALCL), with skin lesions in the same area as the lesion described above and ipsilateral cervical lymph-node involvement at the time of diagnosis. The patient had received chemotherapy (CHOP: cyclophosphamide, doxorubicin, vincristine, and prednisone) and external radiotherapy, with complete remission of the skin lesions and lymph-node involvement.
Examination revealed a 15×12cm plaque affecting the malar, infraocular, and preauricular regions on the left side, extending down to the angle of the mandible. The lesion had poorly defined borders and was formed by the confluence of multiple tumors (Fig. 1).
Histology revealed marked epidermal spongiosis that gave rise to scattered intraepidermal vesicles and intense edema of the papillary dermis (Fig. 2A). A dense lymphocytic proliferation affected the rest of the dermis and extended into the subcutaneous tissue (Fig. 2A). The neoplastic infiltrate was formed by large pleomorphic cells with abundant clear cytoplasm. The nuclei were kidney-shaped or oval with unevenly distributed chromatin and prominent nucleoli (Fig. 2B).

A, Dense lymphocytic infiltrate affecting the full thickness of the dermis and reaching the subcutaneous tissue (hematoxylin-eosin, original magnification x20). B, Lymphoid cells with abundant cytoplasm, marked pleomorphism, mitotic activity, and atypia (hematoxylin-eosin, original magnification, x200). C, Intense positivity for the CD30 marker in almost the entire specimen (x20).
Immunohistochemistry was positive for CD3 and CD30 (Fig. 2C) in 95% of neoplastic cells. CD5 and CD15 expression was focal, with the markers detected in 30% and 20% of the cells, respectively. Ki-67 antigen was positive in 80% to 90% of the neoplastic infiltrate. In addition, 30% of cells were positive for CD4 and 15% were positive for CD8+, while anaplastic lymphoma kinase (ALK) 1, CD56, and epithelial membrane antigen were negative. Genetic study of the T-cell receptor showed monoclonal reordering.
Further tests included an analysis of T-cell populations and immunoglobulin levels, a computed tomography of the head, neck, abdomen and pelvis, and a bone-marrow biopsy. All the results were normal or negative.
These findings led to a diagnosis of cutaneous recurrence of ALK-negative CD30+ anaplastic large-cell lymphoma, and chemotherapy was initiated. The patient showed partial remission after 2 complete cycles of CHOP. One month later the patient suffered a relapse and ESHAP (cytarabine, methylprednisolone, cisplatin, etoposide) therapy was started, resulting in a marked reduction in lesion size by the end of the first cycle.
ALCL can be divided into 2 large groups.1 The first is made up of the primary cutaneous ALCLs that, together with lymphomatoid papulosis, are included in the primary cutaneous CD30+ lymphoproliferative disorders.2 The second group is composed of systemic (nodal) ALCLs, which can be subdivided into ALK positive and negative.
The differential diagnosis of the various types of CD30+ ALCL is complicated but is essential in view of the differences in the prognosis and therapeutic management. ALK protein expression, positive in 80% of systemic ALCLs and negative in almost 100% of primary cutaneous ALCLs, can be useful for differentiating between cutaneous and nodal origins.3–5 The expression of this protein is due to chromosomal translocations, usually t(2;5)(p23;q35).
Translocations that affect the MUM1/IRF4 gene have recently been reported in primary cutaneous ALCL. The specificity of these translocations and their utility as markers to differentiate between the various CD30+ lymphoid proliferations has yet to be established.6
The clinical course may also be helpful in orienting the diagnosis. The 5-year survival of primary cutaneous ALCL is 95%. Primary systemic ALCL, however, has a poorer prognosis, with a 5-year survival of 80% in the ALK-positive subgroup and 30% in ALK-negative cases.3,4,7 There are series of ALK-negative ALCL with cutaneous and nodal involvement at the time of diagnosis, as was the case in our patient, with a survival similar to that of primary cutaneous ALCL.3,8 Given that cutaneous involvement by ALK-negative systemic ALCL is rare, a cutaneous origin with early lymph-node involvement has been suggested to explain the indolent course of these cases.7,8
The treatment of choice for primary systemic ALCL, whether ALK-positive or ALK-negative, is polychemotherapy.9 Treatment for primary cutaneous ALCL, however, includes less aggressive options such as no treatment initially, in the hope of spontaneous resolution, or low-dose methotrexate, local radiation therapy, or surgical resection.10 In our case, given the initial diagnosis of primary nodal ALCL, chemotherapy was chosen to treat the recurrence.
In conclusion, a rare case of ALK-negative ALCL with cutaneous and locoregional lymph-node involvement at the time of diagnosis is reported. Further genetic and immunohistochemical studies are necessary to clarify whether cases such as that of the patient presented in this paper are in actual fact primary cutaneous rather than systemic ALCL.
Please cite this article as: Messeguer F, et al. Inicio cutáneo de un linfoma anaplásico CD 30+ ALK negativo primario sistémico. Actas Dermosifiliogr.2011;102:547-48.


