

El siringoma condroide maligno (SCM), también conocido como tumor mixto maligno cutáneo, es una neoplasia maligna de origen anexial extremadamente rara, con alrededor de 50 casos publicados en la literatura hasta la fecha1. En contraste con su contraparte benigna (tumor mixto benigno), que tiene una predilección por los hombres y por lo general se encuentra en la cabeza, el SCM es más común en las mujeres y se localiza frecuentemente en las extremidades y el tronco2,3. Hasta el momento, solo se han comunicado 10 casos de SCM de compromiso cefálico1–10. A continuación se describe un SCM gigante facial con metástasis a distancia.
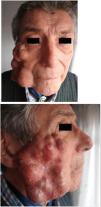
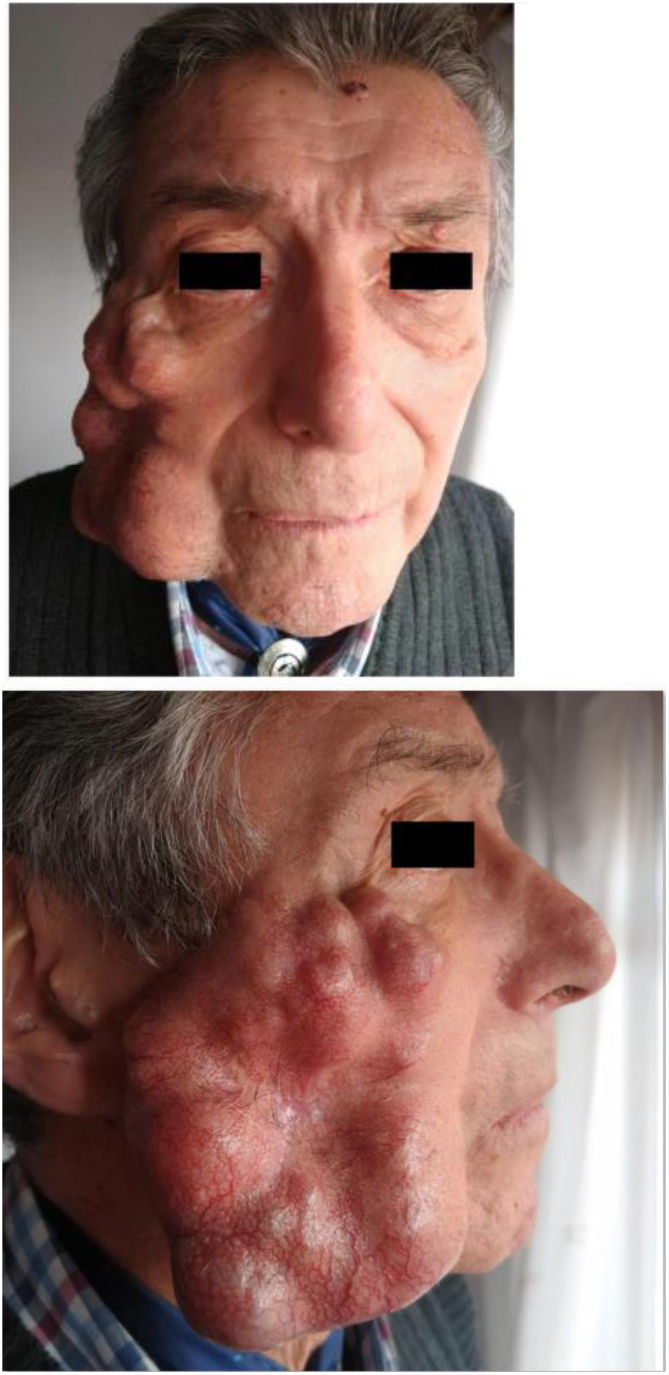
Un hombre de 77 años, con miastenia gravis e hipertensión arterial en tratamiento, presentaba una tumoración indolora en la mejilla derecha de 2 años de evolución. La lesión había crecido abruptamente en los últimos 3 meses. El paciente refería una dificultad para masticar y notaba una disminución de la salivación. En el examen físico se observó un nódulo tumoral subcutáneo de 11×6,7×3,5cm, de color piel normal, con múltiples telangiectasias, multilobulado, firme y adherido a planos profundos, que comprometía la región malar y maxilar inferior derecha de la cara (fig. 1 A y B). Se palpaban adenomegalias axilares homolaterales. Se realizó una tomografía computarizada de la cabeza, que mostró la presencia de una lesión polilobulada heterogénea con unas áreas hipodensas, de 10cm en su diámetro máximo, en la región lateral derecha del macizo facial en contacto con el músculo masetero y con un compromiso de la glándula parótida homolateral. En el estudio histológico de una biopsia incisional, con la tinción de hematoxilina-eosina, se observó una proliferación tumoral de células epiteliales conformando nidos sólidos, cordones y estructuras glanduliformes, destacándose la presencia de focos de necrosis y otros focos hipercelulares con macroanisocariosis, nucléolos evidentes y varias figuras de mitosis (fig. 2). El estroma interpuesto mostró unas áreas predominantemente esclerohialinas, sectores mixoides y otros condroides. Finalmente, la inmunohistoquímica informó CEA, EMA, PS100 y CK positivos, y KI-67 de 25%. Con estos resultados se diagnosticó de un SCM. La pantomografía con emisión de positrones evidenció lesiones nodulares hiperdensas e hipermetabólicas compatibles con metástasis en el cerebro, el hígado, los ganglios axilares derechos y en partes blandas del brazo derecho. El paciente falleció durante el ingreso por causas respiratorias.
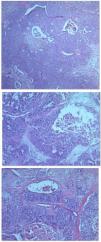
![A) Área sólida del tumor con focos de necrosis y hemorragia (×50 hematoxilina-eosina [H-E]). B) Nidos epiteliales con atipia y necrosis central, rodeados de estroma con aspecto mucinoso (×100 H-E). C) Nidos y cordones epiteliales rodeados de estroma fibroso, con focos de necrosis tumoral (×100 H-E).](https://static.elsevier.es/multimedia/00017310/0000011400000007/v1_202307101051/S0001731022007852/v1_202307101051/es/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w91sAmkCw32Jed9sZf6jzEuDbFpW7G0NfARZs8afh+9K8v8RN+oFy2ZmalFHXVYo6Ar5YDNGfM0MtaqHfFHhhhOBRiIw44x66PJCSKefJgKQSiVAwZUnO0oXz8dBm1OI2dN+ot+o+OdOcr9ysLDMGneGPfk9uYxHlkboSHCklQ59wGUxvK72xeLENHxg1gCec01xwI1exKtelx7G2GGY62wkL58AdYGlLuSSviXv8xxYsxP/dNnIbFefxo3+VPHSotMNx91QFWl60Vp+fTgVUpYE=)
A) Área sólida del tumor con focos de necrosis y hemorragia (×50 hematoxilina-eosina [H-E]). B) Nidos epiteliales con atipia y necrosis central, rodeados de estroma con aspecto mucinoso (×100 H-E). C) Nidos y cordones epiteliales rodeados de estroma fibroso, con focos de necrosis tumoral (×100 H-E).
Descrito por primera vez por Hirsch y Helwig en 1961, el siringoma condroide es un tumor benigno que surge de las glándulas sudoríparas ecrinas o apocrinas4. Es muy infrecuente, representando solo el 0,01% de todas las neoplasias primarias de piel. Se presenta como un nódulo subcutáneo o intradérmico solitario, sólido, indoloro y de crecimiento lento, que afecta cabeza y cuello1,2. Ocurre más comúnmente en hombres de mediana edad. El tamaño del tumor puede variar entre unos 2mm y más de 1cm; aquellos de más de 3cm tienen una mayor probabilidad de malignidad1–3.
A diferencia de su contraparte benigna, el SCM ocurre predominantemente en las mujeres, no tiene predilección relacionada con la edad, y se observa más comúnmente en las extremidades o el torso1,3. Es un tumor de una evolución lenta caracterizada por un período final de crecimiento rápido que puede resultar en destrucción local, metástasis y muerte5. El sitio más común de metástasis a distancia es el pulmón, seguido por el hueso y el cerebro2,4. En el caso comunicado, el tumor ocurrió en un hombre de edad avanzada, se localizó en extremo cefálico y presentó una evolución lenta inicial con una aceleración del crecimiento en el último tiempo. Al momento del diagnóstico, existía invasión local y metástasis en tejido linfático, cerebro, hígado y partes blandas.
El SCM puede desarrollarse de novo o, más raramente, a partir de un siringoma benigno resecado de manera incompleta. La recurrencia de la lesión debe alertar de la posibilidad de malignidad1,2. En este trabajo, el tumor se originó de novo y el paciente no refería antecedentes de lesiones faciales extirpadas.
Aunque algunos autores han sugerido que los traumatismos locales podrían tener un rol en la etiopatogenia, hasta la fecha no se han identificado claramente los factores de riesgo para el siringoma condroide benigno o maligno1,4.
Histológicamente, el siringoma condroide se caracteriza por la presencia de componentes tanto epiteliales como mesenquimales. El componente mesenquimal puede mostrar características mixoides, condroides, osteoides, adiposas o fibrosas y contiene nidos de células epiteliales con una diferenciación glandular ecrina o apocrina1. Los criterios de malignidad incluyen la presencia de atipia citológica, un aumento de la actividad mitótica, la presencia de necrosis tumoral, invasión capsular con compromiso de estructuras profundas y posiblemente nódulos satélites3,7. Los estudios inmunohistoquímicos muestran la presencia simultánea de células luminales CK+, CEA+, EMA+, y células mioepiteliales CK+, PS100 +, vimentina+1. Debido a su presentación clínica poco característica, el diagnóstico de siringoma condroide solo puede hacerse por histopatología.
Los diagnósticos diferenciales incluyen quiste epidérmico, quiste dermoide, quiste pilar, pilomatrixoma, tricoepitelioma, schwannoma, y neurofibroma, entre otros1,2.
El tratamiento del siringoma condroide es la exéresis completa. Es esencial para el control de la enfermedad y para prevenir la recidiva la obtención de un margen quirúrgico libre amplio2,4. Aunque la radioterapia local a menudo no tiene éxito, se ha demostrado que las metástasis óseas responden a la misma. No se ha informado que la quimioterapia combinada sea beneficiosa en pacientes con metástasis2,4.
Debido a la alta tasa de recurrencia de estos tumores (49%), se debe proporcionar un seguimiento cercano a los pacientes y, lo que es más importante, se debe mantener a largo plazo: se han notificado recidivas con diseminación linfática y metástasis a distancia hasta 12 a 20 años después de la extirpación inicial del tumor1.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


