


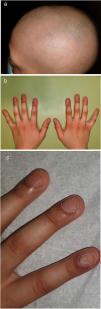
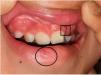
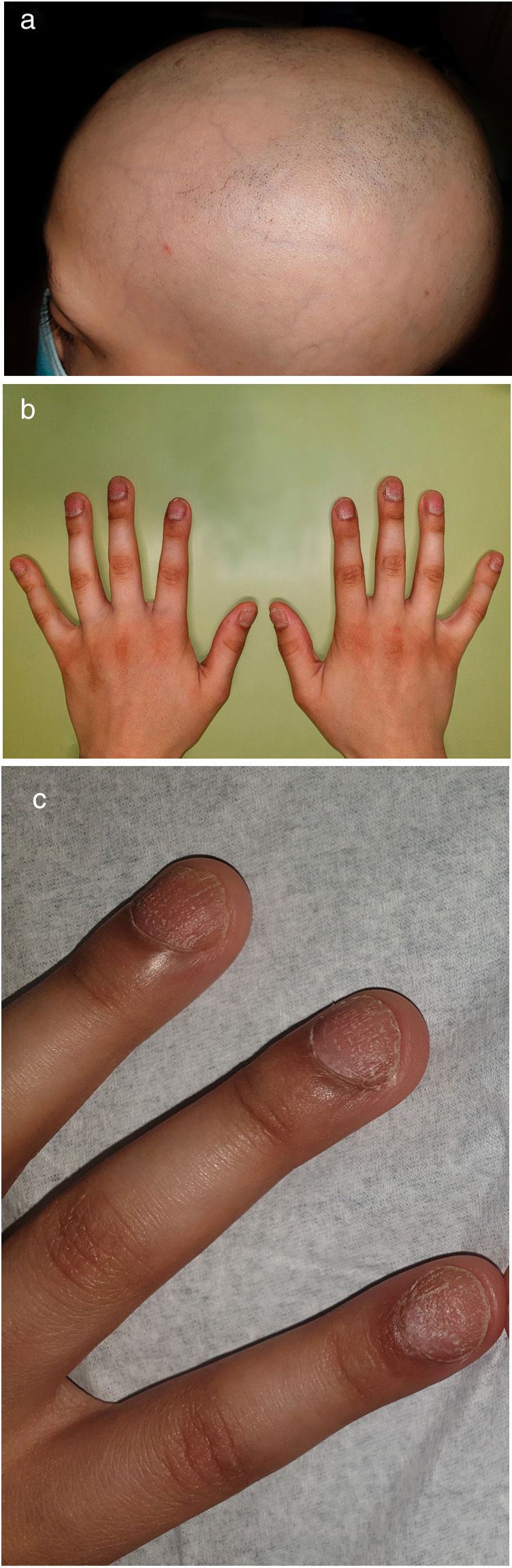
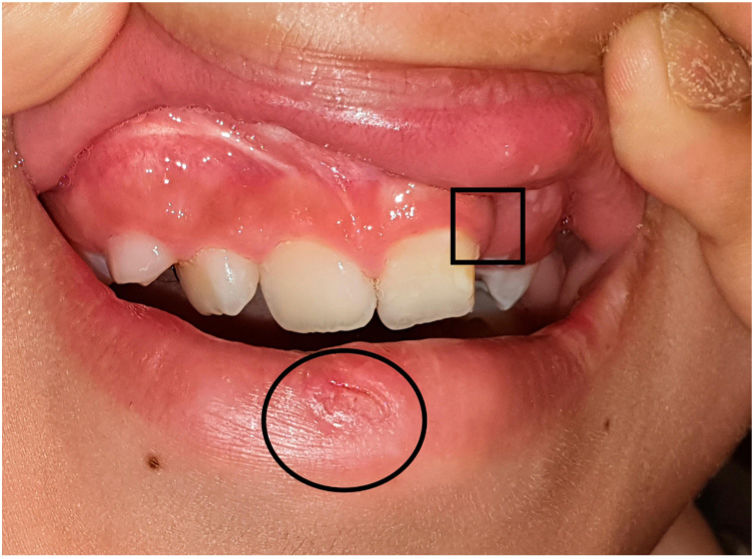
Un niño caucásico de 9 años de edad fue remitido a las consultas externas del Servicio de Dermatología por presentar alopecia y fragilidad de las uñas de las manos, que se habían hecho evidentes desde hacía nueve meses. A la exploración, presentaba alopecia multilocular con extensas áreas confluentes que afectaban al 70% del cuero cabelludo (Fig. 1(a)), a la tricoscopia se observaba la presencia de puntos negros y pelos en signo de exclamación dispersos compatibles con el diagnóstico de alopecia areata. Las cejas también estaban afectadas, especialmente la porción externa. Las uñas de las manos presentaban piqueteados ungueales y traquioniquia en todos los dedos (Fig. 1(b), (c)); las uñas de los pies normales. Además, se observó la presencia de una depresión en el labio inferior, y durante su primera infancia, el paciente tenía el antecedente de haber sido sometido a la corrección quirúrgica de un paladar hendido (Fig. 2). Toda esta clínica en conjunto sugería el diagnóstico de un Síndrome de Van der Woude (SVW). Por lo demás, la historia clínica y la exploración física eran normales. En concreto, no se observaron anomalías en la piel ni en las extremidades (como pterigión poplíteo, sindactilia, cambios en los dedos de los pies) ni malformaciones genitourinarias que pudieran apuntar hacia el síndrome de pterigión poplíteo (SPP), una variante alélica del SVW. Los padres negaron la existencia antecedentes de fisura palatina en otros miembros de la familia. Se pautó una loción de clobetasol de 0,5mg/g y una solución de minoxidil al 5%. El niño presentó una respuesta favorable tras un año de tratamiento (repoblación parcial, con alopecia en el 40% del cuero cabelludo). Las pruebas genéticas fueron positivas para una mutación heterocigota en el gen del factor regulador del interferón 6 (IRF6), concretamente la sustitución de una T por una A en la posición 178, lo que resulta en la sustitución de un triptófano por una arginina en la posición 60 (c.178T>A (p. (Trp60Arg)), una variante que no había sido descrita previamente.
El SVW es un trastorno genético congénito poco frecuente que representa la forma más común de la hendidura orofacial sindrómica, y el 2% de todos los casos de hendidura. Tiene una herencia autosómica dominante de varias mutaciones diferentes en un único gen, el gen IRF6, con alta penetrancia pero con una expresión fenotípica variable. Además de su variada expresividad, el SVW tiene una variante alélica, el Síndrome SPP, que también resulta de mutaciones en el gen IRF6, pero presenta características adicionales además de la presencia de depresiones labiales congénitas y anomalías orofaciales,1 las cuales no se encontraron en nuestro paciente.
El niño fue considerado como el caso índice de la familia. De hecho, el 30-50% de los casos de SVW surgen como mutaciones de novo.1 Sin embargo, no se pudo excluir completamente la presencia de una mutación con penetración reducida, ya que no se realizaron pruebas genéticas en los padres. Se han descrito aproximadamente 170 mutaciones diferentes causantes de la enfermedad en el gen IRF6. Los fenotipos clínicos son el resultado de mutaciones sin sentido, terminaciones, deleciones, inserciones, mutaciones del sitio de empalme, además de otras, pero más del 90% de las mutaciones del IRF6 son cambios sin sentido.1 Además, ya se ha descrito que la misma mutación sin sentido es responsable tanto del SVW como del SPP.1 Sin embargo, como ya se ha mencionado, el fenotipo de nuestro paciente no era compatible con el SPP, y la mutación identificada corresponde a una variante nueva que hasta ahora no se había descrito.
La proteína IRF6 está relacionada con la formación de tejido conjuntivo, concretamente de la cabeza y la cara. El papel del IRF6 en la proliferación epidérmica también se demostró en humanos en estudios histopatológicos, realizados en niños con SVW sometidos a tratamiento quirúrgico del paladar hendido. Sin embargo, a diferencia del SPP que cursa con pterigión poplíteo, hasta ahora no se han descrito alteraciones clínicas de la piel o de los apéndices asociadas al SVW en humanos (excepto un carcinoma de células escamosas asociado a la inflamación crónica de las lesiones labiales).2 Los estudios en ratones también han demostrado que el IRF6 interviene en la proliferación y diferenciación de los queratinocitos, y el fenotipo mutante era muy similar al observado en ratones con epilación repetida (Er), que presentan una caída irregular del cabello.3 Por otra parte, las moléculas de interferón (IFN) (α,β,γ) son factores bien establecidos en la fisiopatología de la alopecia areata4 y se sabe que los factores reguladores del interferón (IRF) desempeñan un papel en la enfermedad impulsada por el IFN.5 Aunque con frecuencia están implicados otros IRF además del IRF6, los efectos pleiotrópicos de estas vías despiertan la atención sobre una posible coocurrencia no casual del SVW y la AA, y debería ser objeto de investigación en el futuro.
Además, los estudios han sugerido un papel cooperativo del p63 y el IRF6 en el desarrollo orofacial de ratones y humanos,6 y las mutaciones en el gen TP63 causan una serie de displasias ectodérmicas que incluyen anomalías del cabello y las uñas. Aunque éstas difieren de la alopecia areata, el vínculo entre el IRF6 y el TP63 debería ser merecedor de un mayor interés.
El presente caso describe una nueva mutación del IRF6, aunque el diseño experimental carece de una prueba funcional directa. También llama la atención sobre una posible asociación no descrita entre un trastorno capilar común y un síndrome genético raro. Así pues, despierta un interés especial por comprender la compleja interacción fisiopatológica de las vías implicadas. Además, pone de manifiesto la importancia de los dermatólogos a la hora de abordar la necesidad de un esfuerzo multidisciplinar de estos síndromes.
Conflictos de interésLos autores declaran no tener ningún conflicto de interés.


